

A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS
- PMID: 31474832
- PMCID: PMC6702328
- DOI: 10.3389/fncel.2019.00346
A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS
Abstract
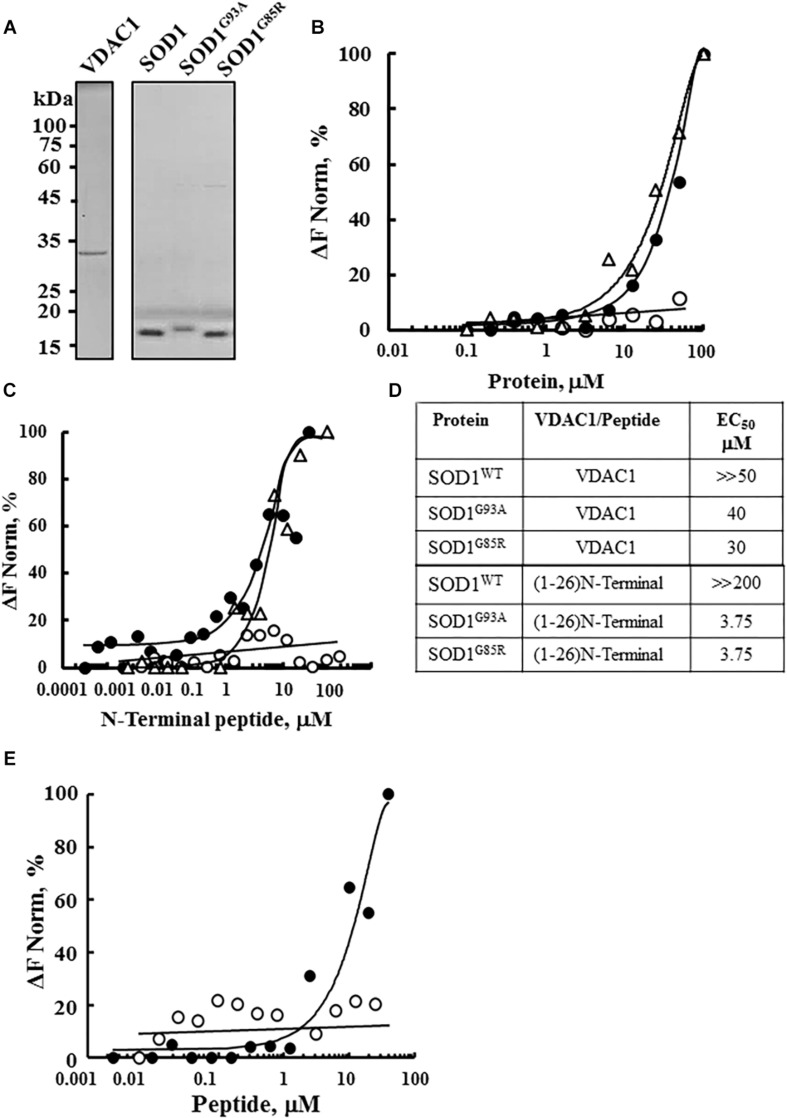
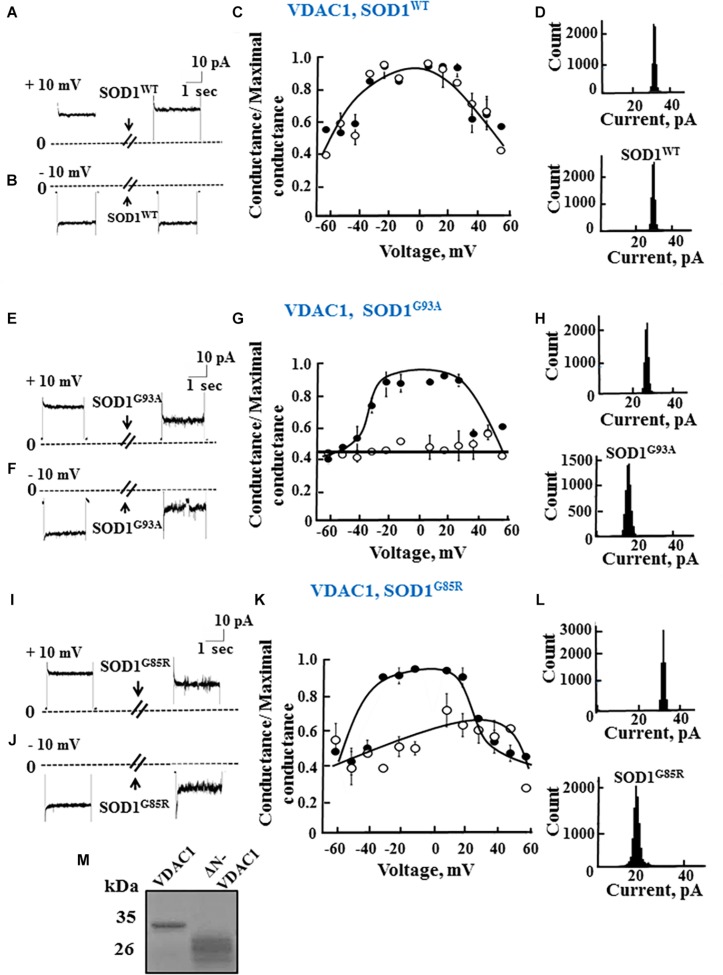
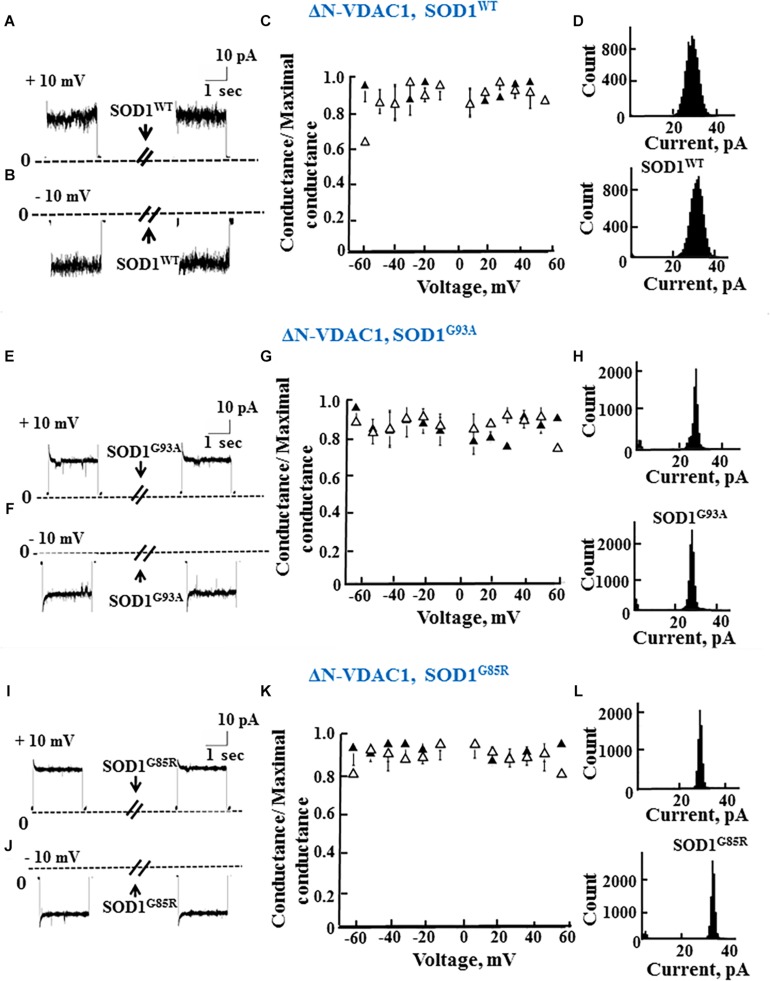
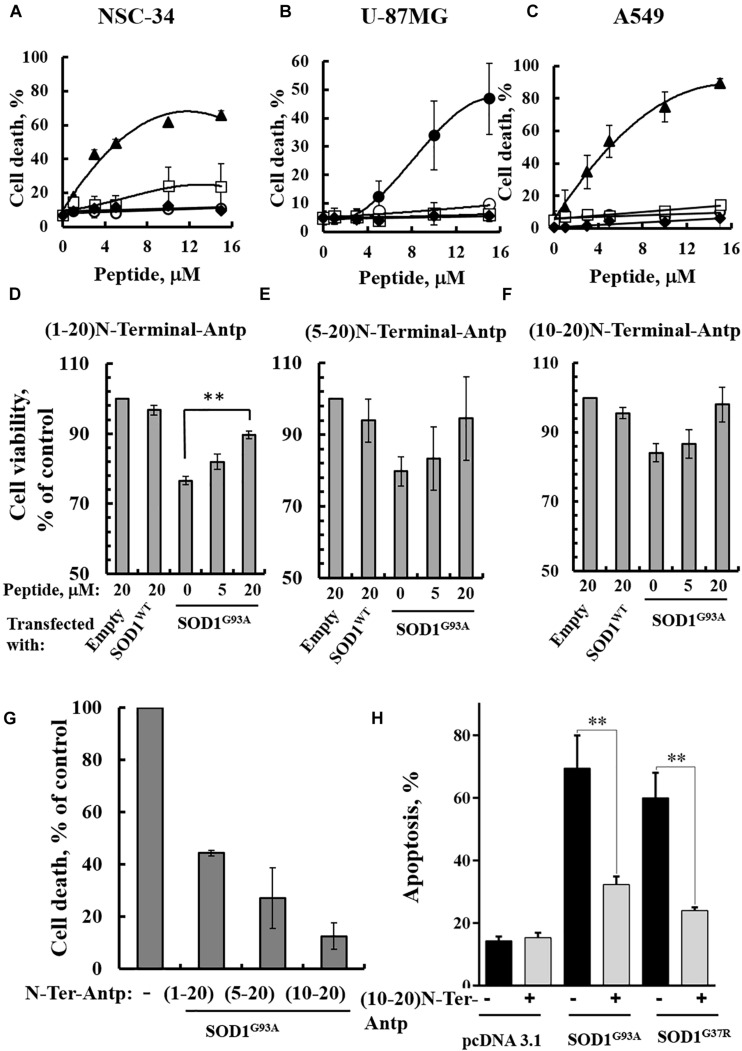
Mutations in superoxide dismutase (SOD1) are the second most common cause of familial amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease caused by the death of motor neurons in the brain and spinal cord. SOD1 neurotoxicity has been attributed to aberrant accumulation of misfolded SOD1, which in its soluble form binds to intracellular organelles, such as mitochondria and ER, disrupting their functions. Here, we demonstrate that mutant SOD1 binds specifically to the N-terminal domain of the voltage-dependent anion channel (VDAC1), an outer mitochondrial membrane protein controlling cell energy, metabolic and survival pathways. Mutant SOD1G93A and SOD1G85R, but not wild type SOD1, directly interact with VDAC1 and reduce its channel conductance. No such interaction with N-terminal-truncated VDAC1 occurs. Moreover, a VDAC1-derived N-terminal peptide inhibited mutant SOD1-induced toxicity. Incubation of motor neuron-like NSC-34 cells expressing mutant SOD1 or mouse embryonic stem cell-derived motor neurons with different VDAC1 N-terminal peptides resulted in enhanced cell survival. Taken together, our results establish a direct link between mutant SOD1 toxicity and the VDAC1 N-terminal domain and suggest that VDAC1 N-terminal peptides targeting mutant SOD1 provide potential new therapeutic strategies for ALS.
Keywords: ALS; N-terminal peptide; VDAC1; misfolded SOD1; mutant SOD1.
Figures
Similar articles
-
Targeting the Mitochondrial Protein VDAC1 as a Potential Therapeutic Strategy in ALS.Int J Mol Sci. 2022 Sep 1;23(17):9946. doi: 10.3390/ijms23179946. Int J Mol Sci. 2022. PMID: 36077343 Free PMC article.
-
Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS.Neuron. 2010 Aug 26;67(4):575-87. doi: 10.1016/j.neuron.2010.07.019. Neuron. 2010. PMID: 20797535 Free PMC article.
-
Hexokinase I N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability.Sci Rep. 2016 Oct 10;6:34802. doi: 10.1038/srep34802. Sci Rep. 2016. PMID: 27721436 Free PMC article.
-
Transgenic mouse model for familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation.Neuropathology. 2001 Mar;21(1):82-92. doi: 10.1046/j.1440-1789.2001.00361.x. Neuropathology. 2001. PMID: 11304046 Review.
-
SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS.Adv Biol Regul. 2016 Jan;60:95-104. doi: 10.1016/j.jbior.2015.10.006. Epub 2015 Oct 31. Adv Biol Regul. 2016. PMID: 26563614 Review.
Cited by
-
Alpha-Synuclein and Mitochondrial Dysfunction in Parkinson's Disease: The Emerging Role of VDAC.Biomolecules. 2021 May 11;11(5):718. doi: 10.3390/biom11050718. Biomolecules. 2021. PMID: 34064816 Free PMC article. Review.
-
Site-specific mitochondrial dysfunction in neurodegeneration.Mitochondrion. 2022 May;64:1-18. doi: 10.1016/j.mito.2022.02.004. Epub 2022 Feb 16. Mitochondrion. 2022. PMID: 35182728 Free PMC article.
-
4-Phenylbutyric Acid (4-PBA) Derivatives Prevent SOD1 Amyloid Aggregation In Vitro with No Effect on Disease Progression in SOD1-ALS Mice.Int J Mol Sci. 2022 Aug 20;23(16):9403. doi: 10.3390/ijms23169403. Int J Mol Sci. 2022. PMID: 36012668 Free PMC article.
-
Empty mesoporous silica particles significantly delay disease progression and extend survival in a mouse model of ALS.Sci Rep. 2020 Nov 26;10(1):20675. doi: 10.1038/s41598-020-77578-x. Sci Rep. 2020. PMID: 33244084 Free PMC article.
-
High-Resolution Respirometry Reveals MPP+ Mitochondrial Toxicity Mechanism in a Cellular Model of Parkinson's Disease.Int J Mol Sci. 2020 Oct 22;21(21):7809. doi: 10.3390/ijms21217809. Int J Mol Sci. 2020. PMID: 33105548 Free PMC article.
References
-
- Abu-Hamad S., Kahn J., Leyton-Jaimes M. F., Rosenblatt J., Israelson A. (2017). Misfolded SOD1 accumulation and mitochondrial association contribute to the selective vulnerability of motor neurons in familial als: correlation to human disease. ACS Chem. Neurosci. 8 2225–2234. 10.1021/acschemneuro.7b00140 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous