

Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) is the Limit
- PMID: 31461845
- PMCID: PMC6747279
- DOI: 10.3390/ijms20174186
Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) is the Limit
Abstract
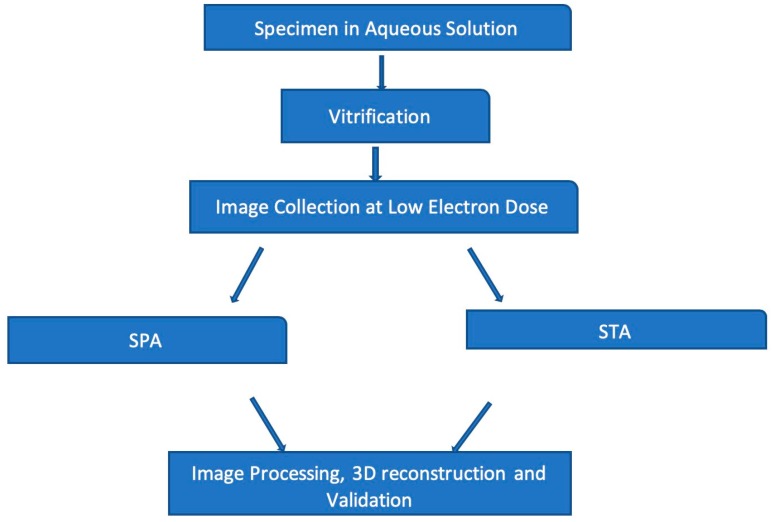
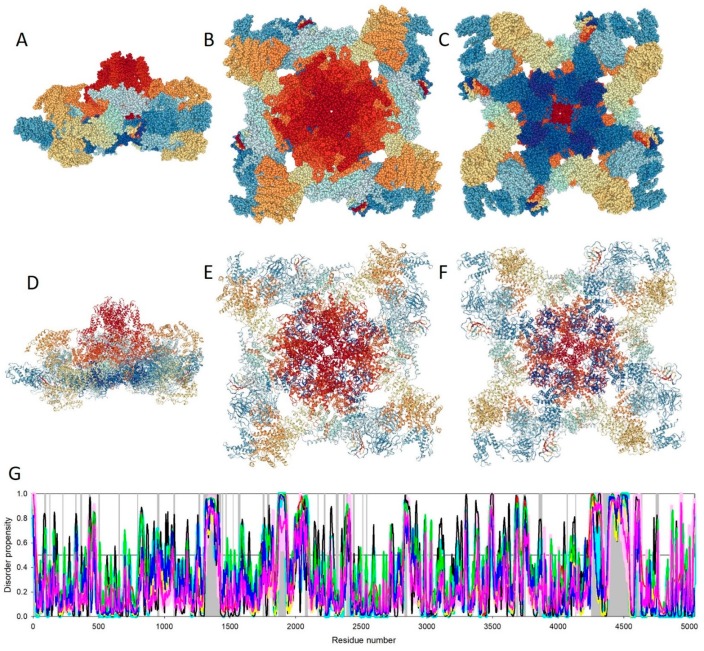
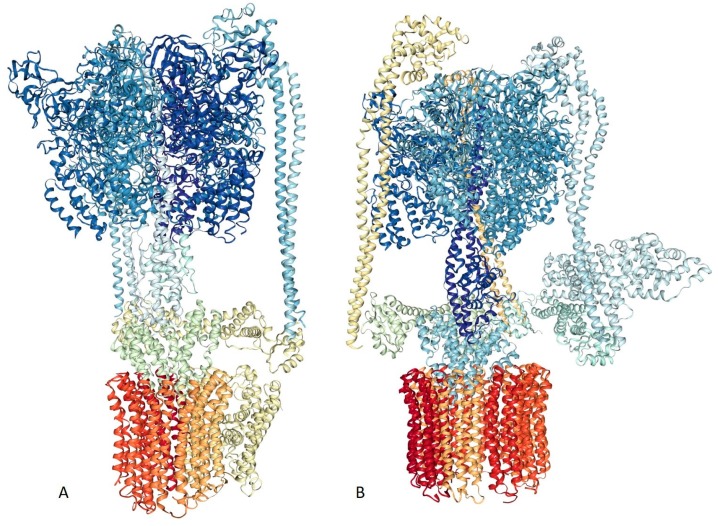
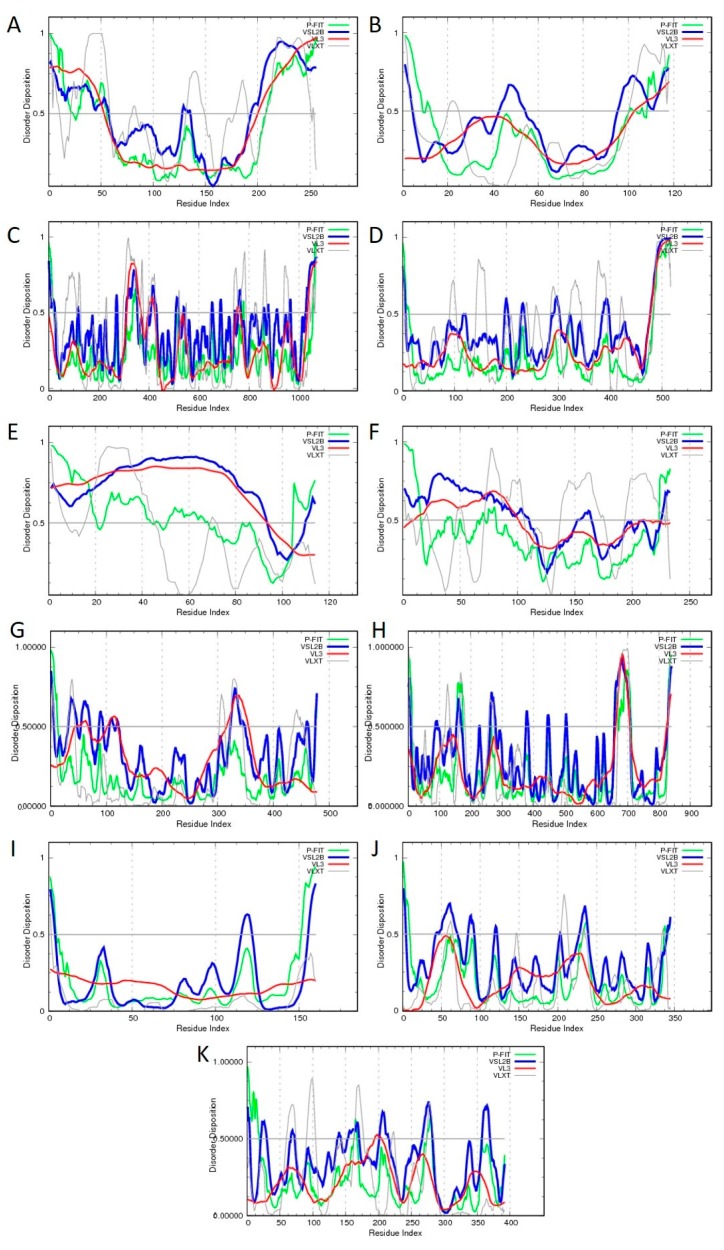
Traditionally, X-ray crystallography and NMR spectroscopy represent major workhorses of structural biologists, with the lion share of protein structures reported in protein data bank (PDB) being generated by these powerful techniques. Despite their wide utilization in protein structure determination, these two techniques have logical limitations, with X-ray crystallography being unsuitable for the analysis of highly dynamic structures and with NMR spectroscopy being restricted to the analysis of relatively small proteins. In recent years, we have witnessed an explosive development of the techniques based on Cryo-electron microscopy (Cryo-EM) for structural characterization of biological molecules. In fact, single-particle Cryo-EM is a special niche as it is a technique of choice for the structural analysis of large, structurally heterogeneous, and dynamic complexes. Here, sub-nanometer atomic resolution can be achieved (i.e., resolution below 10 Å) via single-particle imaging of non-crystalline specimens, with accurate 3D reconstruction being generated based on the computational averaging of multiple 2D projection images of the same particle that was frozen rapidly in solution. We provide here a brief overview of single-particle Cryo-EM and show how Cryo-EM has revolutionized structural investigations of membrane proteins. We also show that the presence of intrinsically disordered or flexible regions in a target protein represents one of the major limitations of this promising technique.
Keywords: 3D-structure; Cryo-electron microscopy; Intrinsically disordered protein; NMR spectroscopy; X-ray crystallography; intrinsically disordered region; protein structure; single-particle Cryo-EM; structural analysis; structural biology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
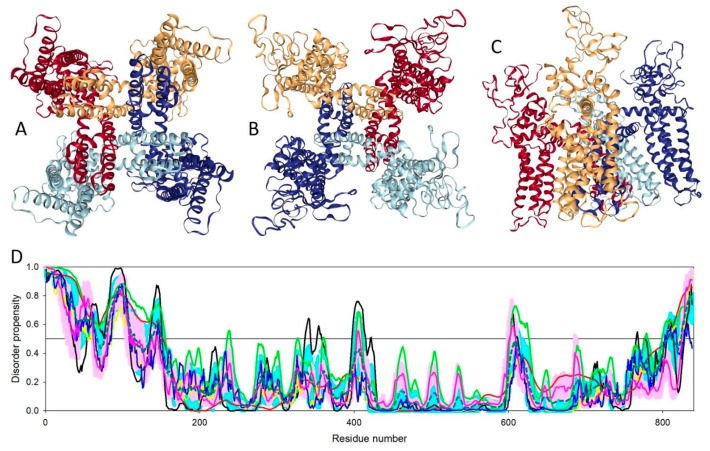
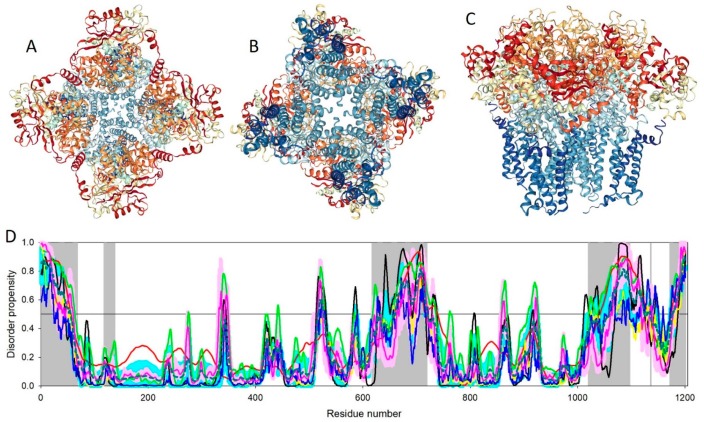
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
