

A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade
- PMID: 31399562
- PMCID: PMC6689050
- DOI: 10.1038/s41467-019-11435-y
A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade
Abstract
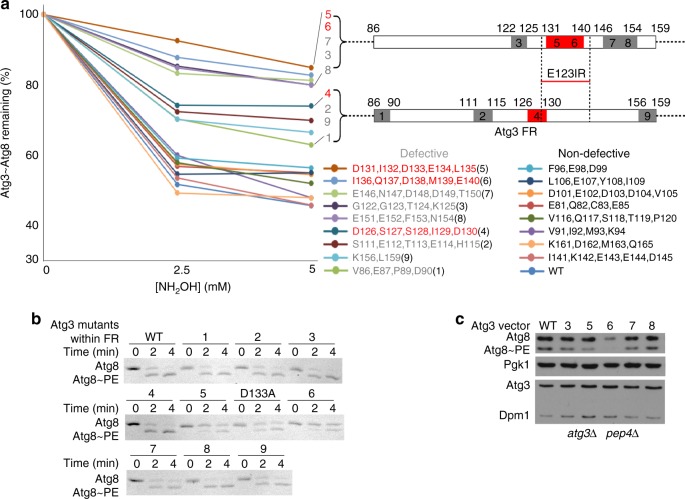
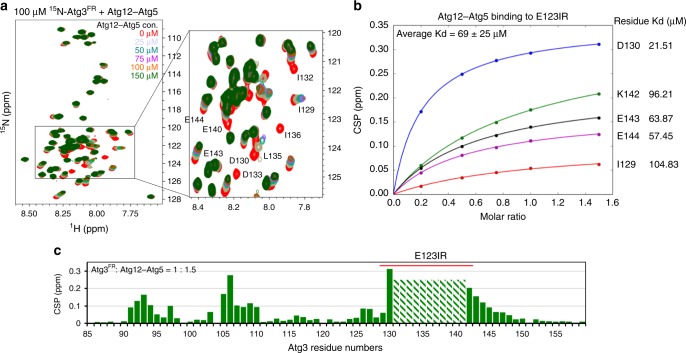
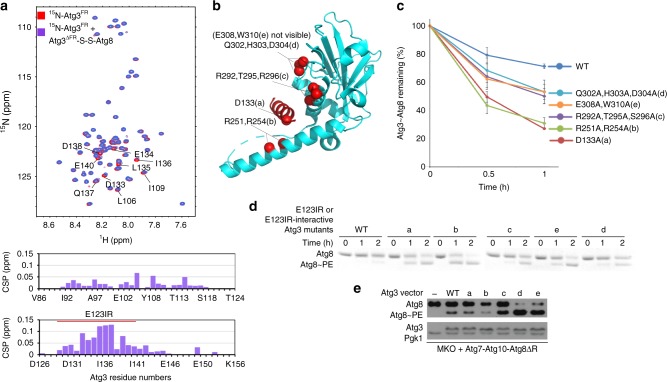
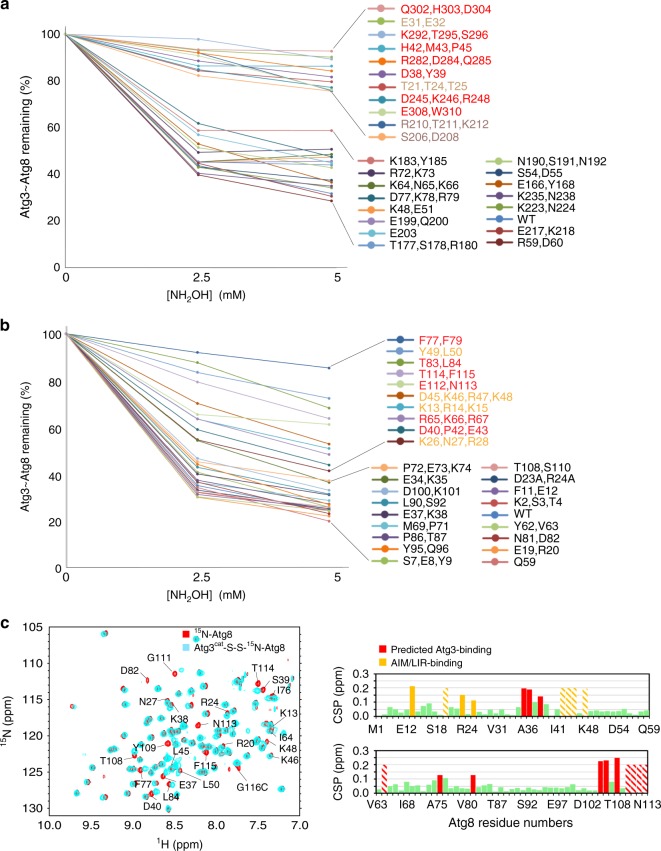
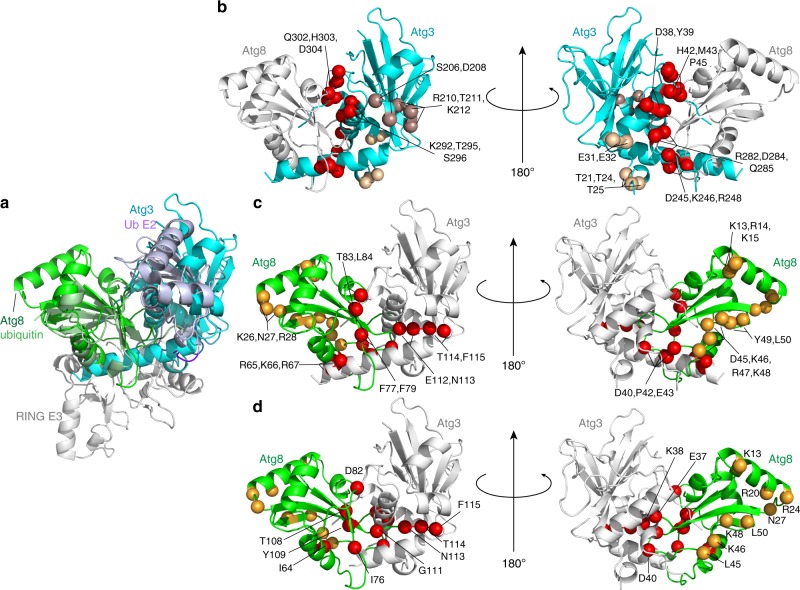
Autophagy depends on the E2 enzyme, Atg3, functioning in a conserved E1-E2-E3 trienzyme cascade that catalyzes lipidation of Atg8-family ubiquitin-like proteins (UBLs). Molecular mechanisms underlying Atg8 lipidation remain poorly understood despite association of Atg3, the E1 Atg7, and the composite E3 Atg12-Atg5-Atg16 with pathologies including cancers, infections and neurodegeneration. Here, studying yeast enzymes, we report that an Atg3 element we term E123IR (E1, E2, and E3-interacting region) is an allosteric switch. NMR, biochemical, crystallographic and genetic data collectively indicate that in the absence of the enzymatic cascade, the Atg3E123IR makes intramolecular interactions restraining Atg3's catalytic loop, while E1 and E3 enzymes directly remove this brace to conformationally activate Atg3 and elicit Atg8 lipidation in vitro and in vivo. We propose that Atg3's E123IR protects the E2~UBL thioester bond from wayward reactivity toward errant nucleophiles, while Atg8 lipidation cascade enzymes induce E2 active site remodeling through an unprecedented mechanism to drive autophagy.
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Allosteric regulation through a switch element in the autophagy E2, Atg3.Autophagy. 2020 Jan;16(1):183-184. doi: 10.1080/15548627.2019.1688550. Epub 2019 Nov 9. Autophagy. 2020. PMID: 31690182 Free PMC article. Review.
-
Binding to E1 and E3 is mutually exclusive for the human autophagy E2 Atg3.Protein Sci. 2013 Dec;22(12):1691-7. doi: 10.1002/pro.2381. Protein Sci. 2013. PMID: 24186333 Free PMC article.
-
Structures of Atg7-Atg3 and Atg7-Atg10 reveal noncanonical mechanisms of E2 recruitment by the autophagy E1.Autophagy. 2013 May;9(5):778-80. doi: 10.4161/auto.23644. Epub 2013 Feb 6. Autophagy. 2013. PMID: 23388412 Free PMC article.
-
Complete set of the Atg8-E1-E2-E3 conjugation machinery forms an interaction web that mediates membrane shaping.Nat Struct Mol Biol. 2024 Jan;31(1):170-178. doi: 10.1038/s41594-023-01132-2. Epub 2023 Dec 6. Nat Struct Mol Biol. 2024. PMID: 38057553
-
In vitro assays of lipidation of Mammalian Atg8 homologs.Curr Protoc Cell Biol. 2014 Sep 2;64:11.20.1-13. doi: 10.1002/0471143030.cb1120s64. Curr Protoc Cell Biol. 2014. PMID: 25181299 Review.
Cited by
-
Effect of ATG12-ATG5-ATG16L1 autophagy E3-like complex on the ability of LC3/GABARAP proteins to induce vesicle tethering and fusion.Cell Mol Life Sci. 2023 Feb 2;80(2):56. doi: 10.1007/s00018-023-04704-z. Cell Mol Life Sci. 2023. PMID: 36729310 Free PMC article.
-
Binding Features and Functions of ATG3.Front Cell Dev Biol. 2021 Jun 21;9:685625. doi: 10.3389/fcell.2021.685625. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34235149 Free PMC article. Review.
-
Unique amphipathic α helix drives membrane insertion and enzymatic activity of ATG3.Sci Adv. 2023 Jun 23;9(25):eadh1281. doi: 10.1126/sciadv.adh1281. Epub 2023 Jun 23. Sci Adv. 2023. PMID: 37352354 Free PMC article.
-
An N-terminal conserved region in human Atg3 couples membrane curvature sensitivity to conjugase activity during autophagy.Nat Commun. 2021 Jan 14;12(1):374. doi: 10.1038/s41467-020-20607-0. Nat Commun. 2021. PMID: 33446636 Free PMC article.
-
Multifaceted membrane interactions of human Atg3 promote LC3-phosphatidylethanolamine conjugation during autophagy.Nat Commun. 2023 Sep 7;14(1):5503. doi: 10.1038/s41467-023-41243-4. Nat Commun. 2023. PMID: 37679347 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
