

Adipocyte JAK2 mediates spontaneous metabolic liver disease and hepatocellular carcinoma
- PMID: 31393852
- PMCID: PMC6777921
- DOI: 10.1172/jci.insight.131310
Adipocyte JAK2 mediates spontaneous metabolic liver disease and hepatocellular carcinoma
Abstract
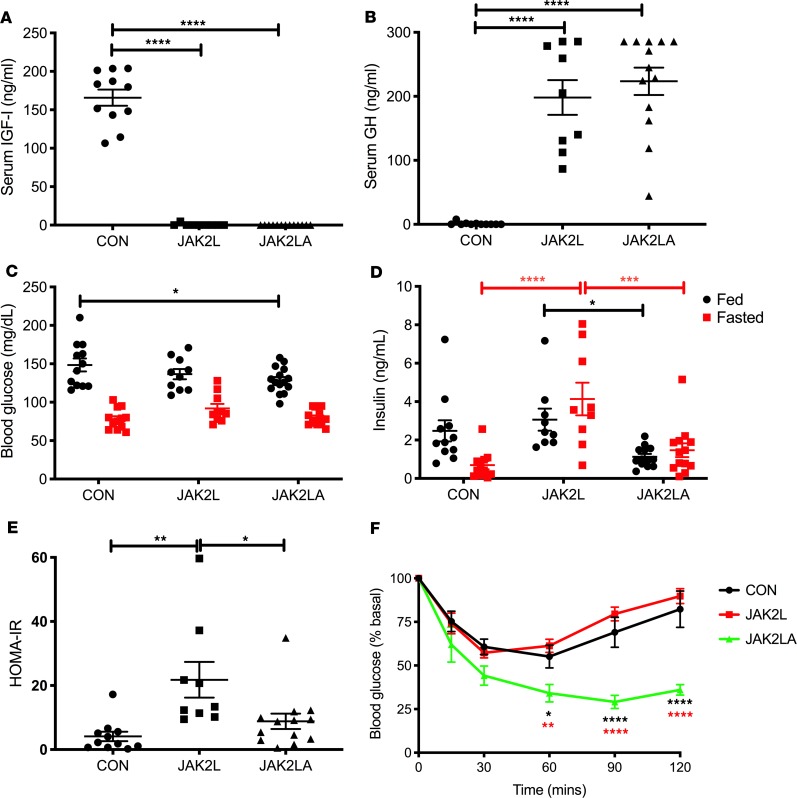
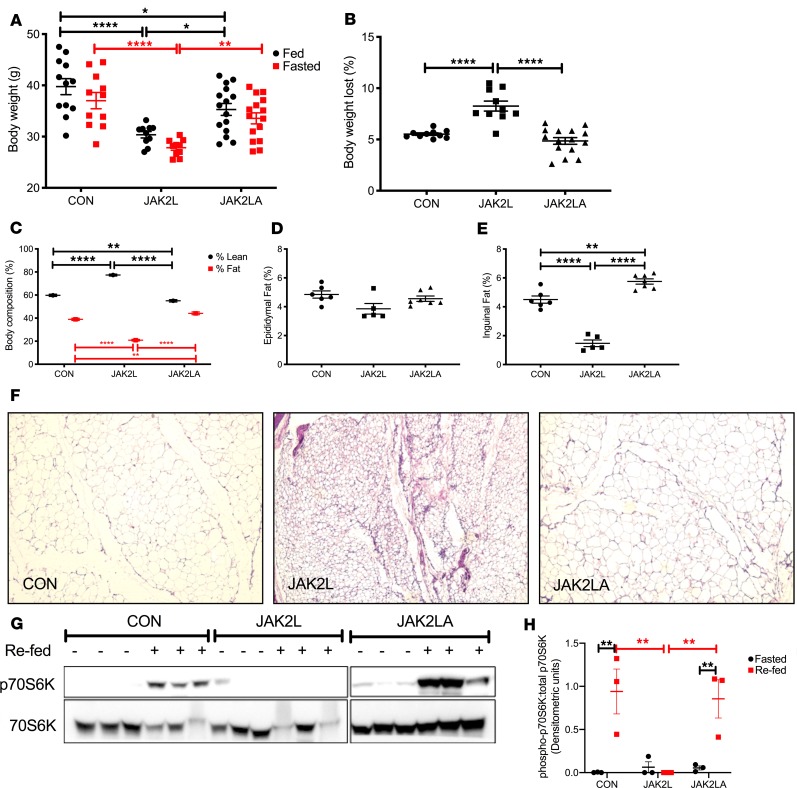
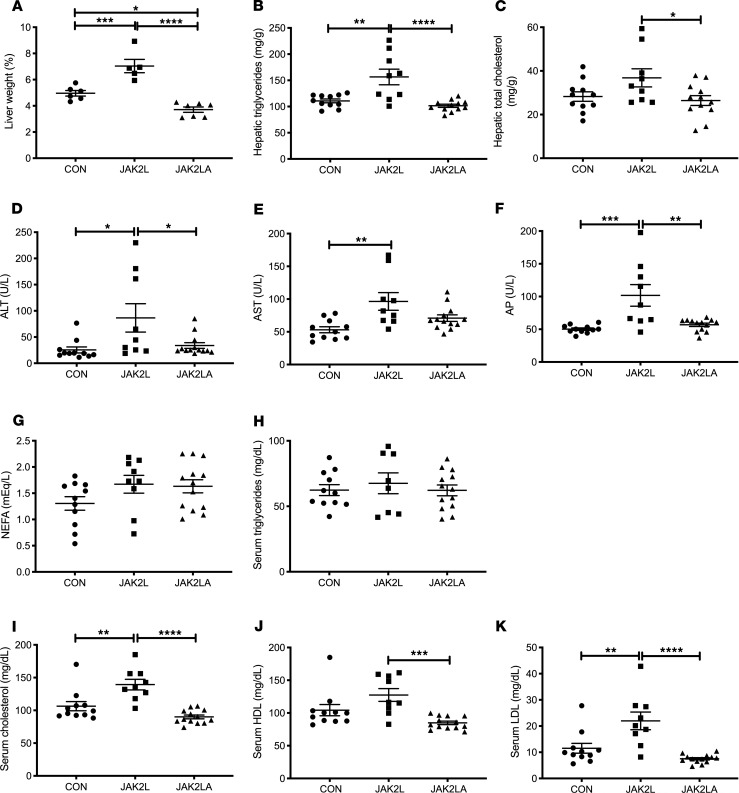
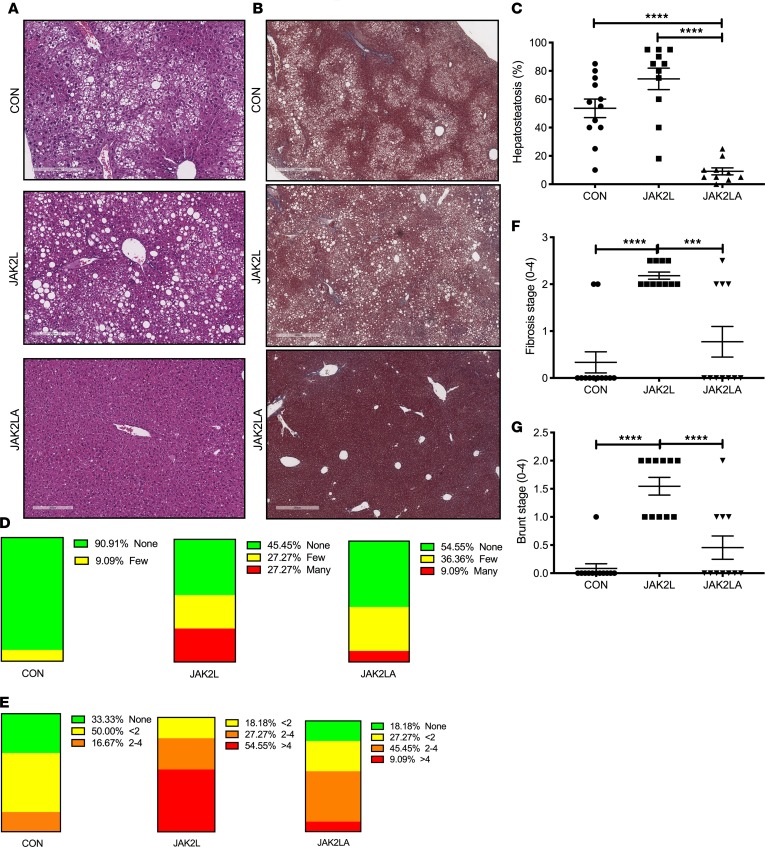
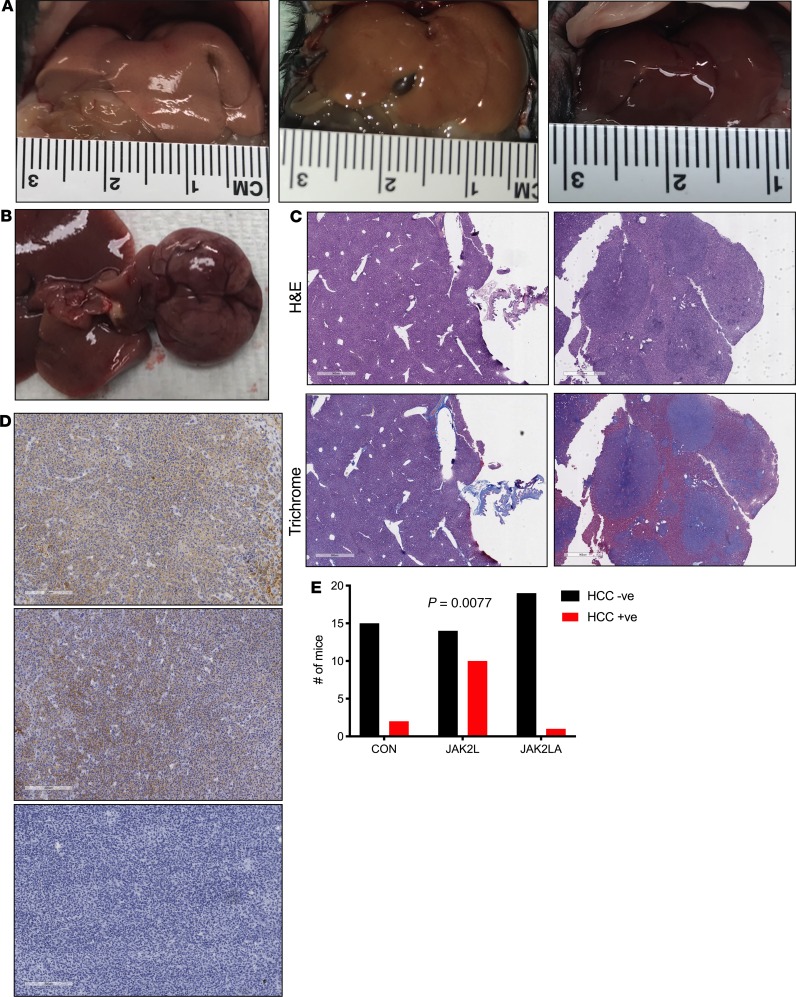
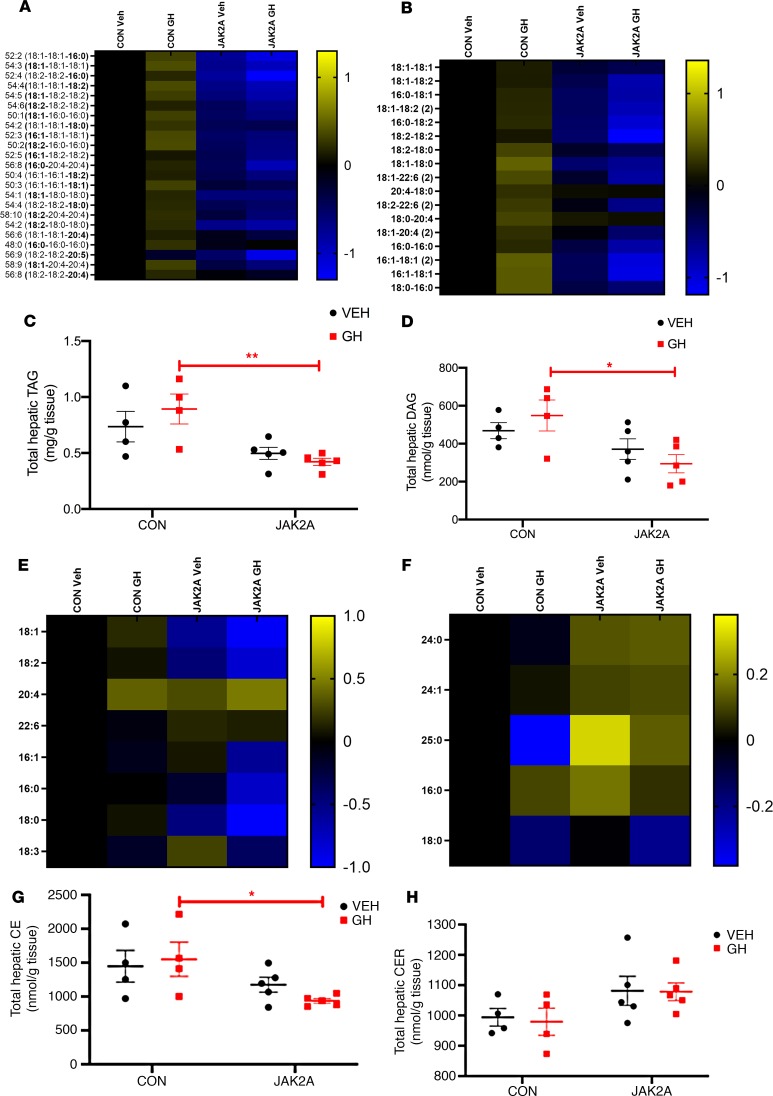
Non-alcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH) are liver manifestations of the metabolic syndrome and can progress to hepatocellular carcinoma (HCC). Loss of Growth Hormone (GH) signaling is reported to predispose to NAFLD and NASH through direct actions on the liver. Here, we report that aged mice lacking hepatocyte Jak2 (JAK2L), an obligate transducer of Growth Hormone (GH) signaling, spontaneously develop the full spectrum of phenotypes found in patients with metabolic liver disease, beginning with insulin resistance and lipodystrophy and manifesting as NAFLD, NASH and even HCC, independent of dietary intervention. Remarkably, insulin resistance, metabolic liver disease, and carcinogenesis are prevented in JAK2L mice via concomitant deletion of adipocyte Jak2 (JAK2LA). Further, we demonstrate that GH increases hepatic lipid burden but does so indirectly via signaling through adipocyte JAK2. Collectively, these data establish adipocytes as the mediator of GH-induced metabolic liver disease and carcinogenesis. In addition, we report a new spontaneous model of NAFLD, NASH, and HCC that recapitulates the natural sequelae of human insulin resistance-associated disease progression. The work presented here suggests a attention be paid towards inhibition of adipocyte GH signaling as a therapeutic target of metabolic liver disease.
Keywords: Hepatology; Insulin; Liver cancer; Metabolism; Mouse models.
Conflict of interest statement
Figures
Similar articles
-
Adipocyte JAK2 Regulates Hepatic Insulin Sensitivity Independently of Body Composition, Liver Lipid Content, and Hepatic Insulin Signaling.Diabetes. 2018 Feb;67(2):208-221. doi: 10.2337/db17-0524. Epub 2017 Dec 4. Diabetes. 2018. PMID: 29203511 Free PMC article.
-
Disruption of JAK2 in adipocytes impairs lipolysis and improves fatty liver in mice with elevated GH.Mol Endocrinol. 2013 Aug;27(8):1333-42. doi: 10.1210/me.2013-1110. Epub 2013 Jun 19. Mol Endocrinol. 2013. PMID: 23782652 Free PMC article.
-
Janus Kinase 2 (JAK2) Dissociates Hepatosteatosis from Hepatocellular Carcinoma in Mice.J Biol Chem. 2017 Mar 3;292(9):3789-3799. doi: 10.1074/jbc.M116.752519. Epub 2017 Jan 18. J Biol Chem. 2017. PMID: 28100771 Free PMC article.
-
Hepatic growth hormone - JAK2 - STAT5 signalling: Metabolic function, non-alcoholic fatty liver disease and hepatocellular carcinoma progression.Cytokine. 2019 Dec;124:154569. doi: 10.1016/j.cyto.2018.10.010. Epub 2018 Oct 30. Cytokine. 2019. PMID: 30389231 Review.
-
Nonalcoholic fatty liver disease and hepatocellular carcinoma.Metabolism. 2016 Aug;65(8):1151-60. doi: 10.1016/j.metabol.2016.01.010. Epub 2016 Jan 23. Metabolism. 2016. PMID: 26907206 Review.
Cited by
-
Loss of Adipocyte STAT5 Confers Increased Depot-Specific Adiposity in Male and Female Mice That Is Not Associated With Altered Adipose Tissue Lipolysis.Front Endocrinol (Lausanne). 2022 Apr 7;13:812802. doi: 10.3389/fendo.2022.812802. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35464049 Free PMC article.
-
Classical and novel GH receptor signaling pathways.Mol Cell Endocrinol. 2020 Dec 1;518:110999. doi: 10.1016/j.mce.2020.110999. Epub 2020 Aug 22. Mol Cell Endocrinol. 2020. PMID: 32835785 Free PMC article. Review.
-
GH directly inhibits steatosis and liver injury in a sex-dependent and IGF1-independent manner.J Endocrinol. 2021 Jan;248(1):31-44. doi: 10.1530/JOE-20-0326. J Endocrinol. 2021. PMID: 33112796 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
