

αKlotho-FGF23 interactions and their role in kidney disease: a molecular insight
- PMID: 31350618
- PMCID: PMC11105488
- DOI: 10.1007/s00018-019-03241-y
αKlotho-FGF23 interactions and their role in kidney disease: a molecular insight
Abstract
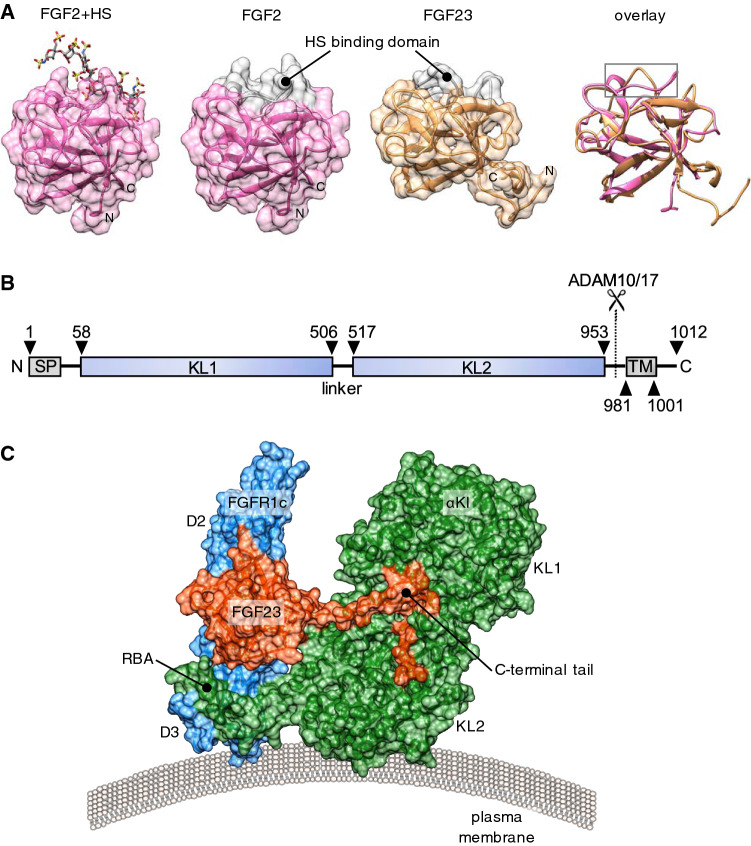
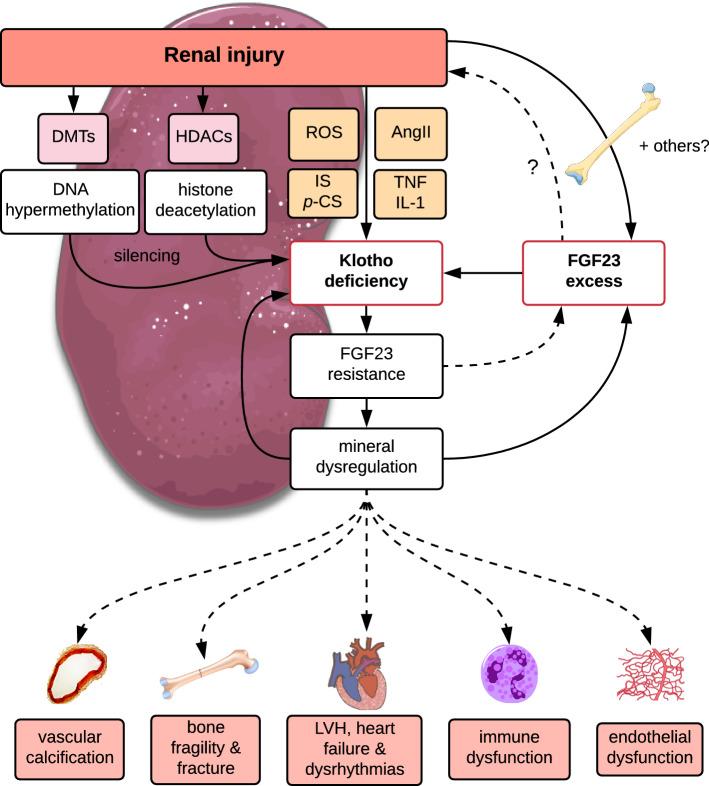
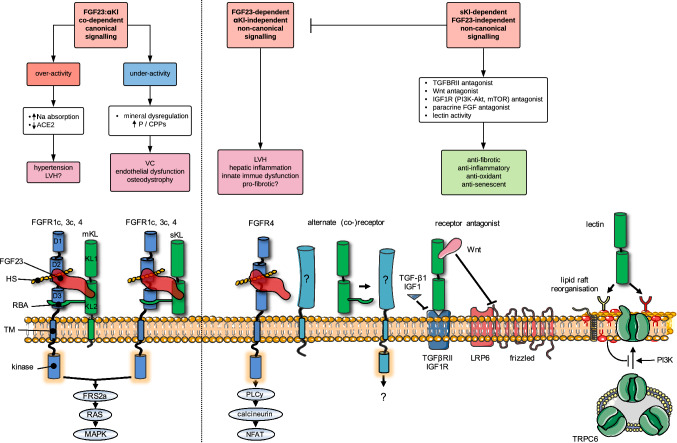
Following the serendipitous discovery of the ageing suppressor, αKlotho (αKl), several decades ago, a growing body of evidence has defined a pivotal role for its various forms in multiple aspects of vertebrate physiology and pathology. The transmembrane form of αKl serves as a co-receptor for the osteocyte-derived mineral regulator, fibroblast growth factor (FGF)23, principally in the renal tubules. However, compelling data also suggest that circulating soluble forms of αKl, derived from the same source, may have independent homeostatic functions either as a hormone, glycan-cleaving enzyme or lectin. Chronic kidney disease (CKD) is of particular interest as disruption of the FGF23-αKl axis is an early and common feature of disease manifesting in markedly deficient αKl expression, but FGF23 excess. Here we critically discuss recent findings in αKl biology that conflict with the view that soluble αKl has substantive functions independent of FGF23 signalling. Although the issue of whether soluble αKl can act without FGF23 has yet to be resolved, we explore the potential significance of these contrary findings in the context of CKD and highlight how this endocrine pathway represents a promising target for novel anti-ageing therapeutics.
Keywords: Cardiovascular disease; Crystallography; Fibroblast growth factor; Kidney disease; Klotho proteins structural biology; Phosphate; Receptors; Therapeutics.
Conflict of interest statement
The authors declare they have no relevant conflicts of interest.
Figures
Similar articles
-
Novel functions of circulating Klotho.Bone. 2017 Jul;100:36-40. doi: 10.1016/j.bone.2016.11.025. Epub 2016 Nov 23. Bone. 2017. PMID: 27890549 Free PMC article. Review.
-
Chronic Hyperphosphatemia and Vascular Calcification Are Reduced by Stable Delivery of Soluble Klotho.J Am Soc Nephrol. 2017 Apr;28(4):1162-1174. doi: 10.1681/ASN.2015111266. Epub 2016 Nov 11. J Am Soc Nephrol. 2017. PMID: 27837149 Free PMC article.
-
New insights into the FGF23-Klotho axis.Semin Nephrol. 2014 Nov;34(6):586-97. doi: 10.1016/j.semnephrol.2014.09.005. Semin Nephrol. 2014. PMID: 25498378 Review.
-
FGF23 induced left ventricular hypertrophy mediated by FGFR4 signaling in the myocardium is attenuated by soluble Klotho in mice.J Mol Cell Cardiol. 2020 Jan;138:66-74. doi: 10.1016/j.yjmcc.2019.11.149. Epub 2019 Nov 21. J Mol Cell Cardiol. 2020. PMID: 31758962 Free PMC article.
-
FGF23-αKlotho as a paradigm for a kidney-bone network.Bone. 2017 Jul;100:4-18. doi: 10.1016/j.bone.2016.11.013. Epub 2016 Nov 12. Bone. 2017. PMID: 27847255 Review.
Cited by
-
Vascular Calcification in Chronic Kidney Disease: An Update and Perspective.Aging Dis. 2022 Jun 1;13(3):673-697. doi: 10.14336/AD.2021.1024. eCollection 2022 Jun. Aging Dis. 2022. PMID: 35656113 Free PMC article.
-
Klotho Pathways, Myelination Disorders, Neurodegenerative Diseases, and Epigenetic Drugs.Biores Open Access. 2020 Mar 31;9(1):94-105. doi: 10.1089/biores.2020.0004. eCollection 2020. Biores Open Access. 2020. PMID: 32257625 Free PMC article. Review.
-
Klotho, Aging, and the Failing Kidney.Front Endocrinol (Lausanne). 2020 Aug 27;11:560. doi: 10.3389/fendo.2020.00560. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 32982966 Free PMC article. Review.
-
New concepts in regulation and function of the FGF23.Clin Exp Med. 2023 Aug;23(4):1055-1066. doi: 10.1007/s10238-022-00844-x. Epub 2022 Jun 16. Clin Exp Med. 2023. PMID: 35708778 Review.
-
Hypoxia-Induced Kidney Injury in Newborn Rats.Toxics. 2023 Mar 11;11(3):260. doi: 10.3390/toxics11030260. Toxics. 2023. PMID: 36977025 Free PMC article.
References
-
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
