

The peptide hormone adropin regulates signal transduction pathways controlling hepatic glucose metabolism in a mouse model of diet-induced obesity
- PMID: 31324719
- PMCID: PMC6737218
- DOI: 10.1074/jbc.RA119.008967
The peptide hormone adropin regulates signal transduction pathways controlling hepatic glucose metabolism in a mouse model of diet-induced obesity
Abstract
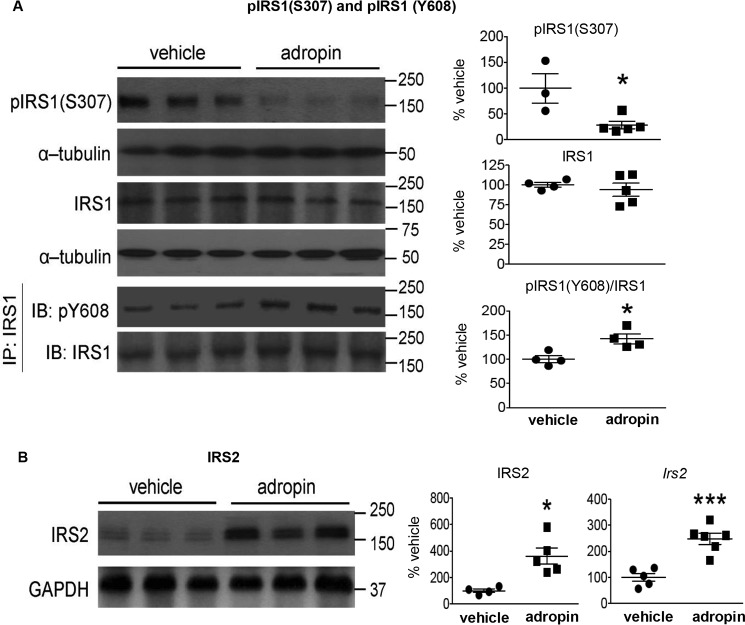
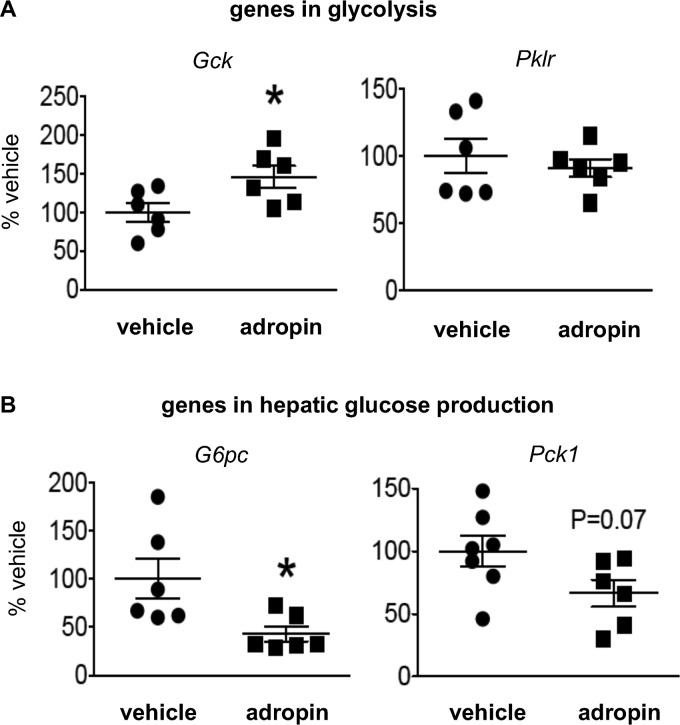
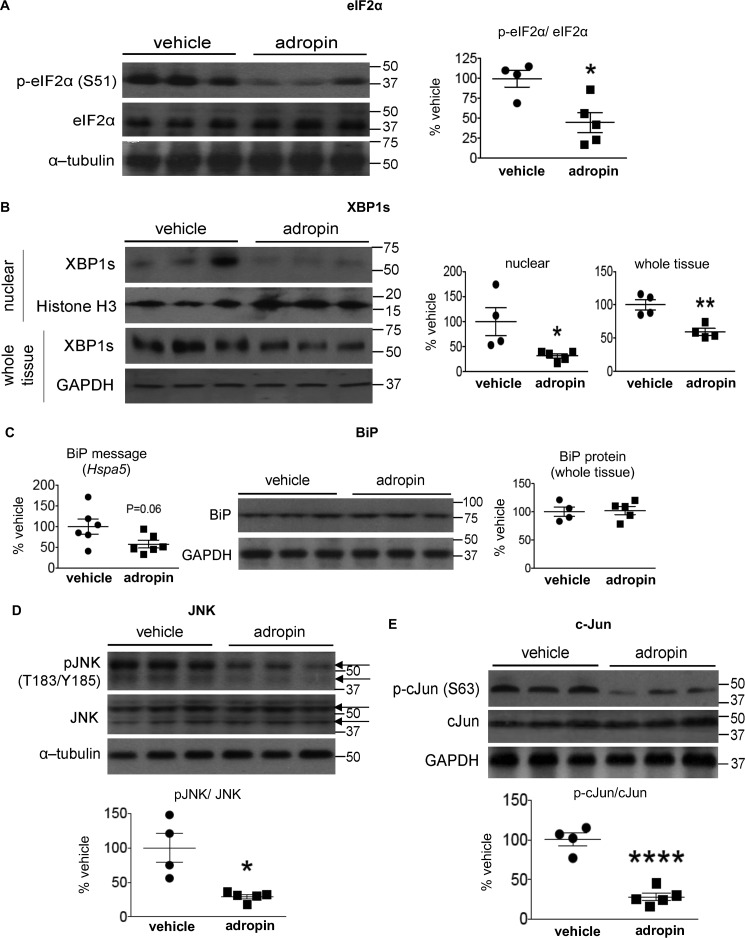
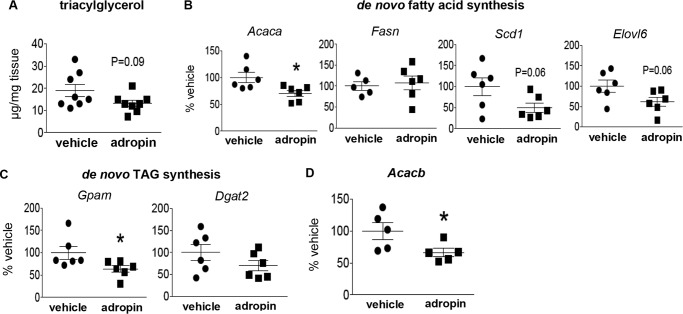
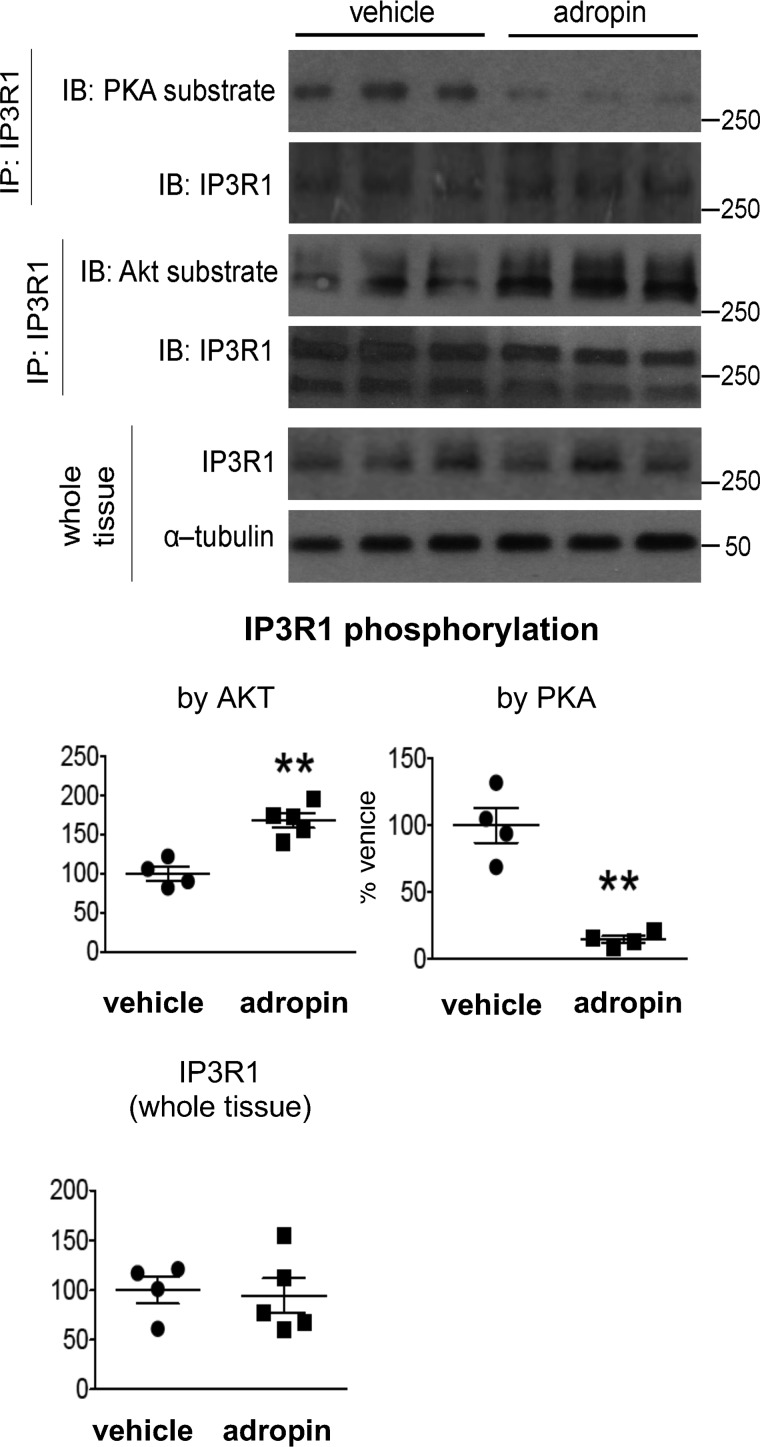
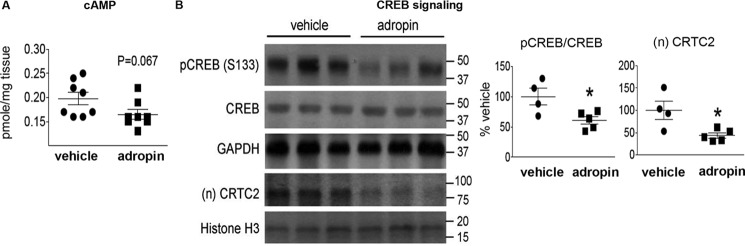
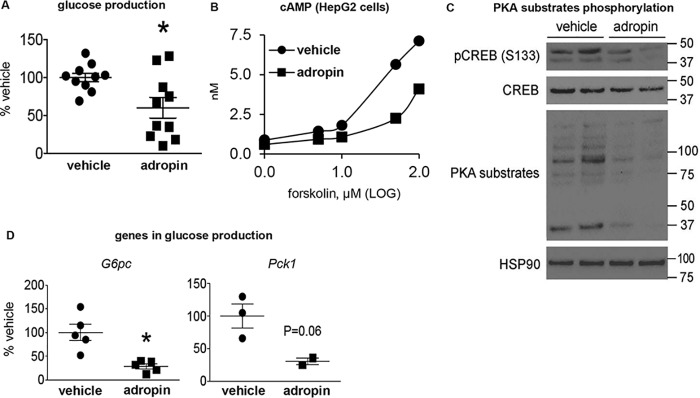
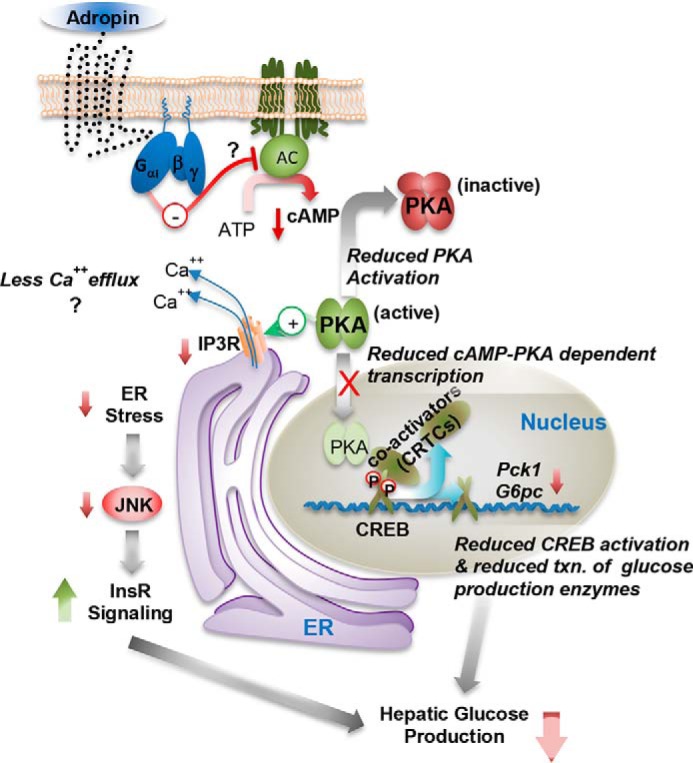
The peptide hormone adropin regulates energy metabolism in skeletal muscle and plays important roles in the regulation of metabolic homeostasis. Besides muscle, the liver has an essential role in regulating glucose homeostasis. Previous studies have reported that treatment of diet-induced obese (DIO) male mice with adropin34-76 (the putative secreted domain) reduces fasting blood glucose independently of body weight changes, suggesting that adropin suppresses glucose production in the liver. Here, we explored the molecular mechanisms underlying adropin's effects on hepatic glucose metabolism in DIO mice. Male DIO B6 mice maintained on a high-fat diet received five intraperitoneal injections of adropin34-76 (450 nmol/kg/injection) over a 48-h period. We found that adropin34-76 enhances major intracellular signaling activities in the liver that are involved in insulin-mediated regulation of glucose homeostasis. Moreover, treatment with adropin34-76 alleviated endoplasmic reticulum stress responses and reduced activity of c-Jun N-terminal kinase in the liver, explaining the enhanced activities of hepatic insulin signaling pathways observed with adropin34-76 treatment. Furthermore, adropin34-76 suppressed cAMP activated protein kinase A (PKA) activities, resulting in reduced phosphorylation of inositol trisphosphate receptor, which mediates endoplasmic reticulum calcium efflux, and of cAMP-responsive element-binding protein, a key transcription factor in hepatic regulation of glucose metabolism. Adropin34-76 directly affected liver metabolism, decreasing glucose production and reducing PKA-mediated phosphorylation in primary mouse hepatocytes in vitro Our findings indicate that major hepatic signaling pathways contribute to the improved glycemic control achieved with adropin34-76 treatment in situations of obesity.
Keywords: adropin; c-Jun N-terminal kinase (JNK); endoplasmic reticulum stress (ER stress); energy metabolism; gluconeogenesis; insulin receptor substrate 1 (IRS1); insulin resistance; lipogenesis; protein kinase A (PKA); type 2 diabetes.
© 2019 Gao et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures


Similar articles
-
Hepatocyte expression of the micropeptide adropin regulates the liver fasting response and is enhanced by caloric restriction.J Biol Chem. 2020 Oct 2;295(40):13753-13768. doi: 10.1074/jbc.RA120.014381. Epub 2020 Jul 29. J Biol Chem. 2020. PMID: 32727846 Free PMC article.
-
Adropin regulates hepatic glucose production via PP2A/AMPK pathway in insulin-resistant hepatocytes.FASEB J. 2020 Aug;34(8):10056-10072. doi: 10.1096/fj.202000115RR. Epub 2020 Jun 24. FASEB J. 2020. PMID: 32579277
-
Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance.Mol Metab. 2015 Jan 17;4(4):310-24. doi: 10.1016/j.molmet.2015.01.005. eCollection 2015 Apr. Mol Metab. 2015. PMID: 25830094 Free PMC article.
-
Adropin's Role in Energy Homeostasis and Metabolic Disorders.Int J Mol Sci. 2022 Jul 28;23(15):8318. doi: 10.3390/ijms23158318. Int J Mol Sci. 2022. PMID: 35955453 Free PMC article. Review.
-
Human Protein Kinases and Obesity.Adv Exp Med Biol. 2017;960:111-134. doi: 10.1007/978-3-319-48382-5_5. Adv Exp Med Biol. 2017. PMID: 28585197 Review.
Cited by
-
Role of angiopoietin-like protein 3 in sugar-induced dyslipidemia in rhesus macaques: suppression by fish oil or RNAi.J Lipid Res. 2020 Mar;61(3):376-386. doi: 10.1194/jlr.RA119000423. Epub 2020 Jan 9. J Lipid Res. 2020. PMID: 31919051 Free PMC article.
-
The Usefulness of Diagnostic Panels Based on Circulating Adipocytokines/Regulatory Peptides, Renal Function Tests, Insulin Resistance Indicators and Lipid-Carbohydrate Metabolism Parameters in Diagnosis and Prognosis of Type 2 Diabetes Mellitus with Obesity.Biomolecules. 2020 Sep 9;10(9):1304. doi: 10.3390/biom10091304. Biomolecules. 2020. PMID: 32917052 Free PMC article.
-
Circulating levels of adropin and diabetes: a systematic review and meta-analysis of observational studies.BMC Endocr Disord. 2023 Apr 7;23(1):73. doi: 10.1186/s12902-023-01327-0. BMC Endocr Disord. 2023. PMID: 37029398 Free PMC article.
-
Low circulating adropin concentrations predict increased risk of cognitive decline in community-dwelling older adults.Geroscience. 2024 Feb;46(1):897-911. doi: 10.1007/s11357-023-00824-3. Epub 2023 May 26. Geroscience. 2024. PMID: 37233882 Free PMC article.
-
ERα-Dependent Regulation of Adropin Predicts Sex Differences in Liver Homeostasis during High-Fat Diet.Nutrients. 2022 Aug 10;14(16):3262. doi: 10.3390/nu14163262. Nutrients. 2022. PMID: 36014766 Free PMC article.
References
-
- Gao S., McMillan R. P., Jacas J., Zhu Q., Li X., Kumar G. K., Casals N., Hegardt F. G., Robbins P. D., Lopaschuk G. D., Hulver M. W., and Butler A. A. (2014) Regulation of substrate oxidation preferences in muscle by the peptide hormone adropin. Diabetes 63, 3242–3252 10.2337/db14-0388 - DOI - PMC - PubMed
-
- Kumar K. G., Trevaskis J. L., Lam D. D., Sutton G. M., Koza R. A., Chouljenko V. N., Kousoulas K. G., Rogers P. M., Kesterson R. A., Thearle M., Ferrante A. W. Jr., Mynatt R. L., Burris T. P., Dong J. Z., Halem H. A., et al. (2008) Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 8, 468–481 10.1016/j.cmet.2008.10.011 - DOI - PMC - PubMed
-
- Butler A. A., Tam C. S., Stanhope K. L., Wolfe B. M., Ali M. R., O'Keeffe M., St-Onge M. P., Ravussin E., and Havel P. J. (2012) Low circulating adropin concentrations with obesity and aging correlate with risk factors for metabolic disease and increase after gastric bypass surgery in humans. J. Clin. Endocrinol. Metab. 97, 3783–3791 10.1210/jc.2012-2194 - DOI - PMC - PubMed
-
- Butler A. A., Zhang J., Price C. A., Stevens J. R., Graham J. L., Stanhope K. L., King S., Krauss R. M., Bremer A. A., and Havel P. J. (2019) Low plasma adropin concentrations increase risks of weight gain and metabolic dysregulation in response to a high-sugar diet in male nonhuman primates. J. Biol. Chem. 294, 9706–9719 10.1074/jbc.RA119.007528 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
