

Hypoxia inducible factor 1α in vascular smooth muscle cells promotes angiotensin II-induced vascular remodeling via activation of CCL7-mediated macrophage recruitment
- PMID: 31320613
- PMCID: PMC6639417
- DOI: 10.1038/s41419-019-1757-0
Hypoxia inducible factor 1α in vascular smooth muscle cells promotes angiotensin II-induced vascular remodeling via activation of CCL7-mediated macrophage recruitment
Abstract
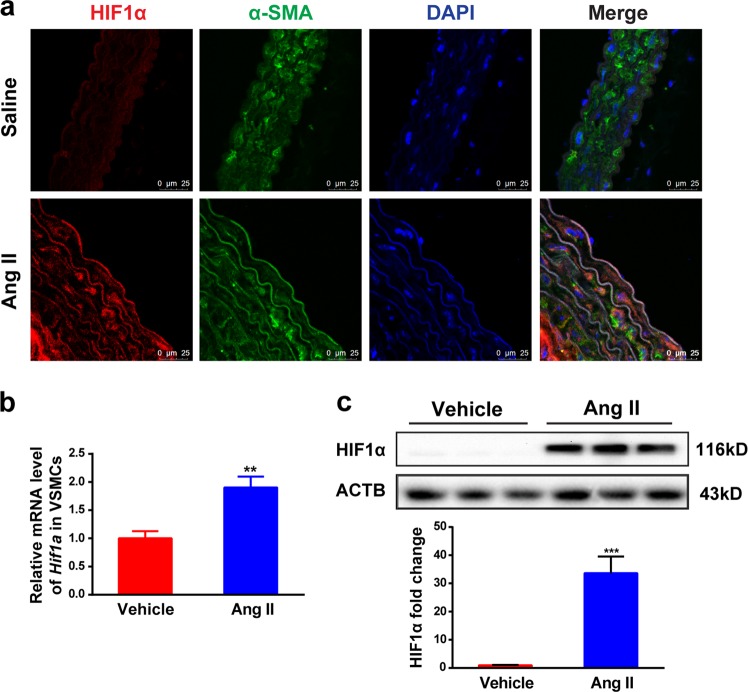
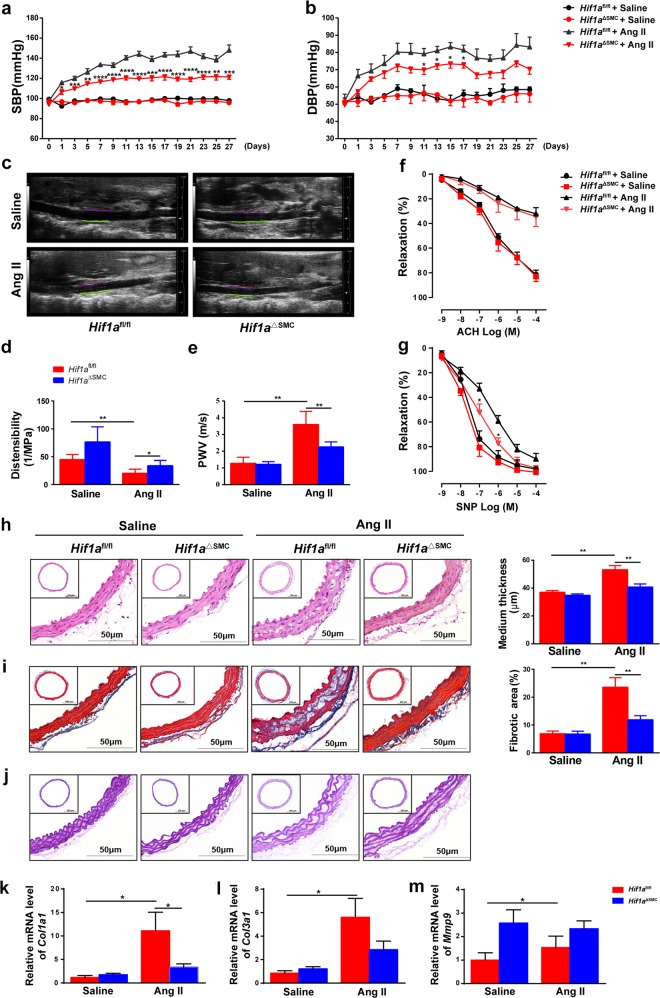
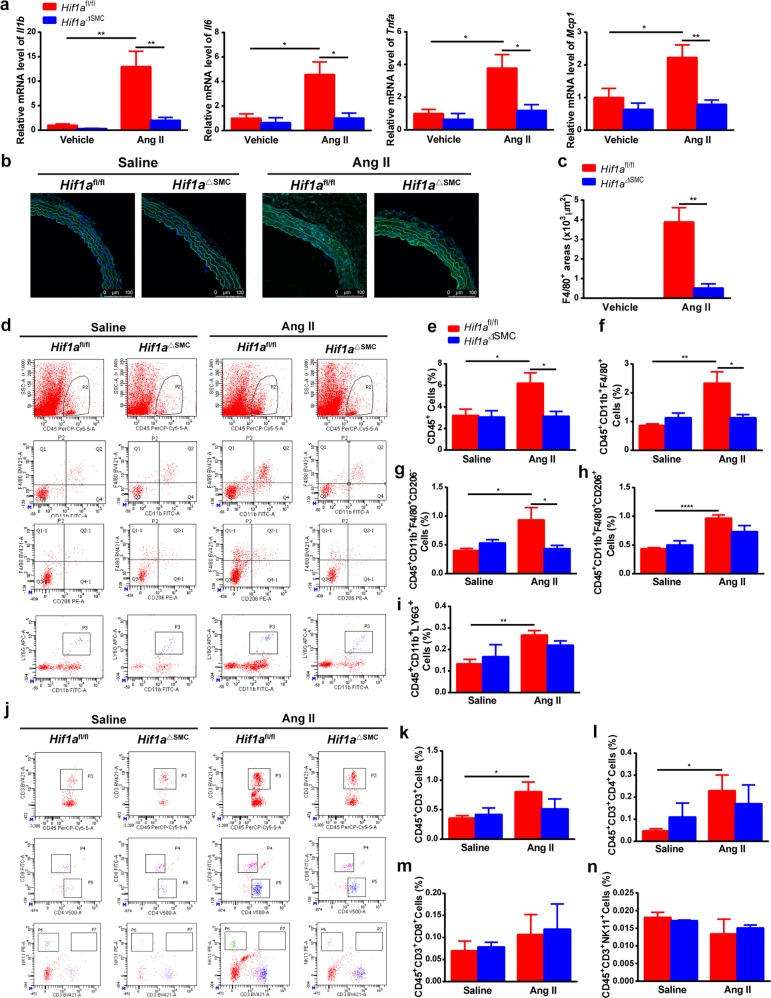
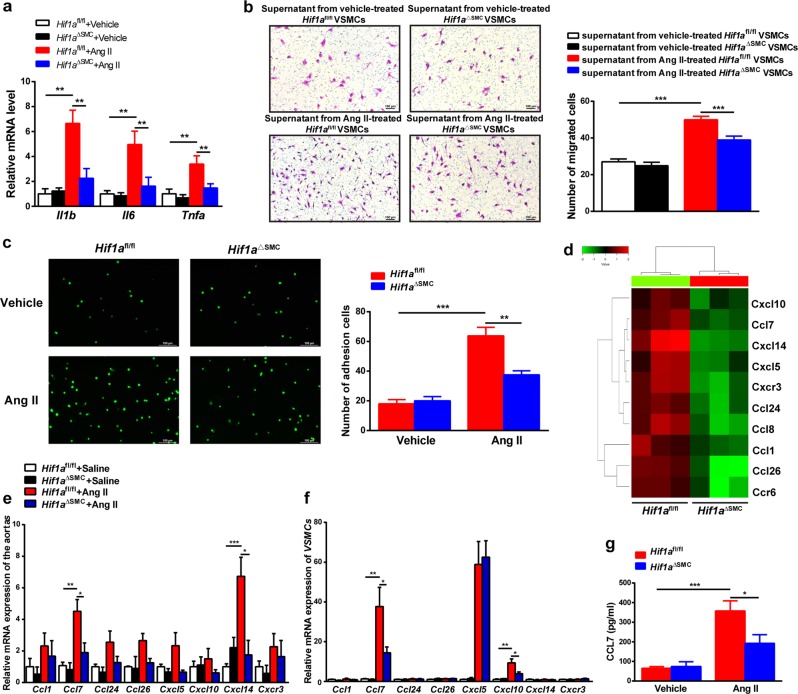
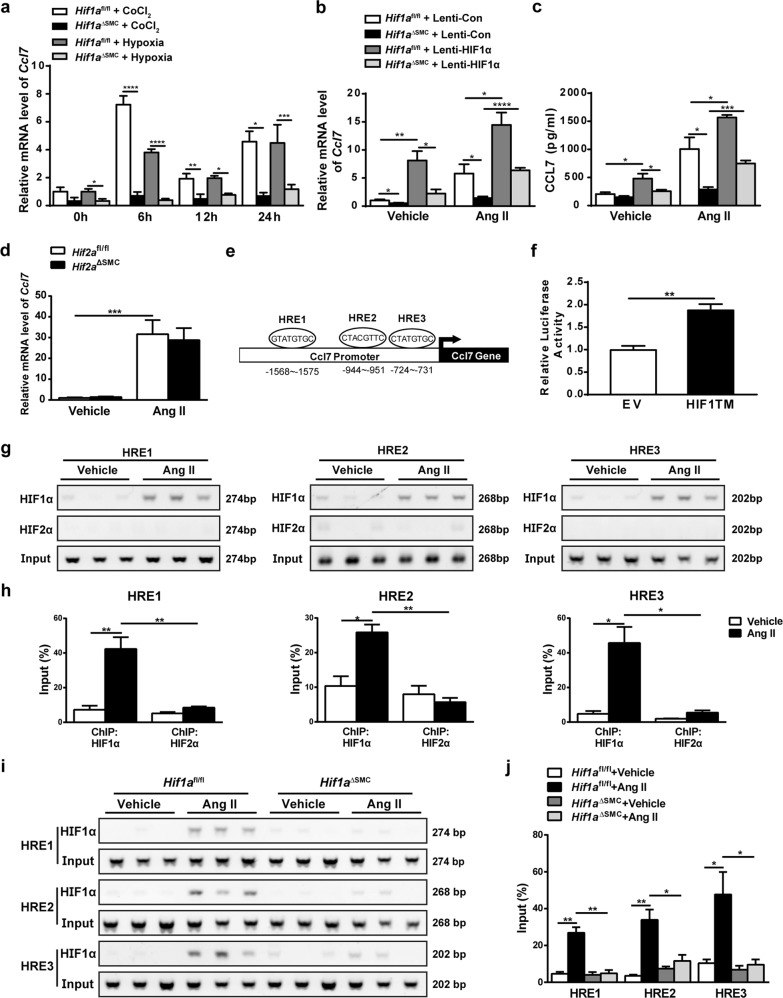
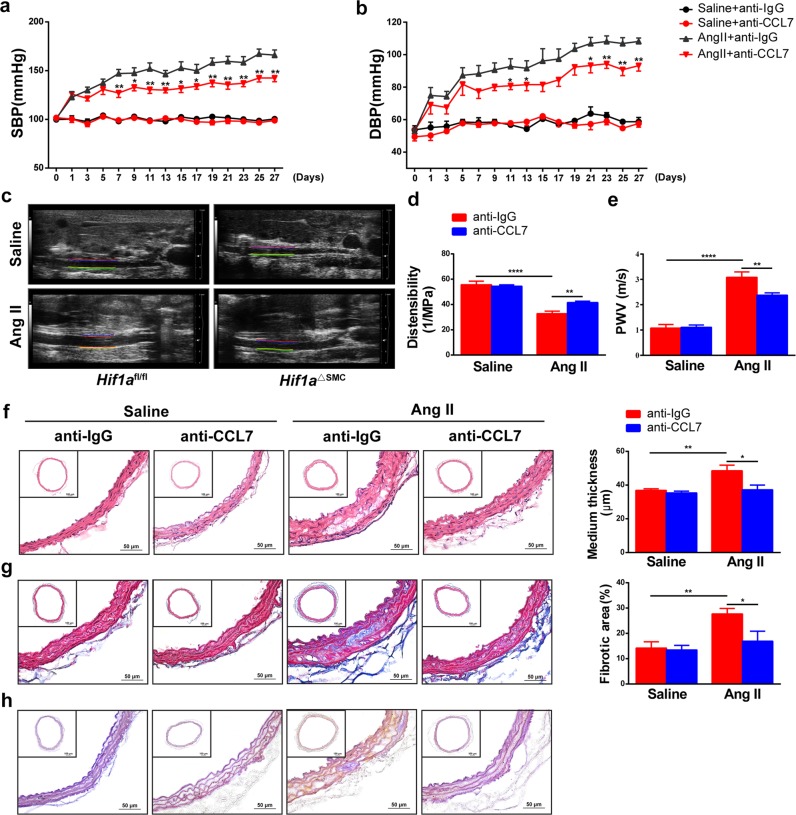
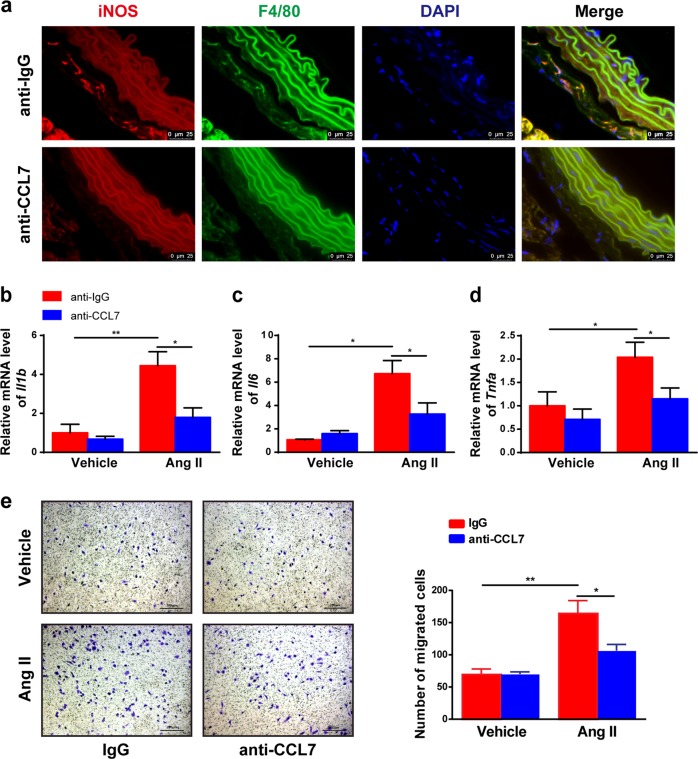
The process of vascular remodeling is associated with increased hypoxia. However, the contribution of hypoxia-inducible factor 1α (HIF1α), the key transcription factor mediating cellular hypoxic responses, to vascular remodeling is established, but not completely understood. In the angiotensin II (Ang II)-induced vascular remodeling model, HIF1α was increased and activated in vascular smooth muscle cells (VSMCs). Selective genetic disruption of Hif1a in VSMCs markedly ameliorated Ang II-induced vascular remodeling, as revealed by decreased blood pressure, aortic thickness, collagen deposition, inflammation, and aortic stiffness. VSMC Hif1a deficiency also specifically suppressed Ang II-induced infiltration of CD45+CD11b+F4/80+CD206- M1 macrophages into the vessel. Mechanistically, HIF1α deficiency in VSMCs dramatically suppressed the expression of CCL7, a chemokine critical for macrophage recruitment. Bioinformatic analysis and chromatin immunoprecipitation assays revealed three functional hypoxia-response elements in the Ccl7 promoter, indicating that Ccl7 is a direct HIF1α target gene. Blocking CCL7 with antibody in vivo alleviated Ang II-induced hypertension and vascular remodeling, coincident with decreased macrophage infiltration. This study provides direct evidence that HIF1α activation in VSMCs exacerbates Ang II-induced macrophage infiltration and resultant vascular remodeling via its target gene Ccl7, and thus may serve as a potential therapeutic target for remodeling-related vascular disease.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Smooth muscle-specific TMEM16A expression protects against angiotensin II-induced cerebrovascular remodeling via suppressing extracellular matrix deposition.J Mol Cell Cardiol. 2019 Sep;134:131-143. doi: 10.1016/j.yjmcc.2019.07.002. Epub 2019 Jul 10. J Mol Cell Cardiol. 2019. PMID: 31301303
-
Smooth muscle cell-specific Hif-1α deficiency suppresses angiotensin II-induced vascular remodelling in mice.Cardiovasc Res. 2014 Jun 1;102(3):460-8. doi: 10.1093/cvr/cvu061. Epub 2014 Mar 12. Cardiovasc Res. 2014. PMID: 24623277
-
HIF-1α knockdown attenuates phenotypic transformation and oxidative stress induced by high salt in human aortic vascular smooth muscle cells.Sci Rep. 2024 Nov 15;14(1):28100. doi: 10.1038/s41598-024-79892-0. Sci Rep. 2024. PMID: 39543255 Free PMC article.
-
Hypoxia-Inducible Factor-1α: The Master Regulator of Endothelial Cell Senescence in Vascular Aging.Cells. 2020 Jan 13;9(1):195. doi: 10.3390/cells9010195. Cells. 2020. PMID: 31941032 Free PMC article. Review.
-
HIF1α and metabolic reprogramming in inflammation.J Clin Invest. 2016 Oct 3;126(10):3699-3707. doi: 10.1172/JCI84431. Epub 2016 Aug 29. J Clin Invest. 2016. PMID: 27571407 Free PMC article. Review.
Cited by
-
Comparative Proteomic Analysis of tPVAT during Ang II Infusion.Biomedicines. 2021 Dec 2;9(12):1820. doi: 10.3390/biomedicines9121820. Biomedicines. 2021. PMID: 34944635 Free PMC article.
-
Adipocyte death triggers a pro-inflammatory response and induces metabolic activation of resident macrophages.Cell Death Dis. 2021 Jun 5;12(6):579. doi: 10.1038/s41419-021-03872-9. Cell Death Dis. 2021. PMID: 34091595 Free PMC article.
-
Immunometabolism at the Heart of Cardiovascular Disease.JACC Basic Transl Sci. 2023 Apr 26;8(7):884-904. doi: 10.1016/j.jacbts.2022.12.010. eCollection 2023 Jul. JACC Basic Transl Sci. 2023. PMID: 37547069 Free PMC article. Review.
-
Morin Inhibits Dox-Induced Vascular Inflammation By Regulating PTEN/AKT/NF-κB Pathway.Inflammation. 2022 Dec;45(6):2406-2418. doi: 10.1007/s10753-022-01701-5. Epub 2022 Jun 15. Inflammation. 2022. PMID: 35705830 Free PMC article.
-
CC chemokines Modulate Immune responses in Pulmonary Hypertension.J Adv Res. 2024 Sep;63:171-186. doi: 10.1016/j.jare.2023.10.015. Epub 2023 Nov 4. J Adv Res. 2024. PMID: 37926143 Free PMC article. Review.
References
-
- Collaborators, G. B. D. R. F. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1659–1724. doi: 10.1016/S0140-6736(16)31679-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
