

Acetaminophen hepatotoxicity: A mitochondrial perspective
- PMID: 31307587
- PMCID: PMC7350903
- DOI: 10.1016/bs.apha.2019.01.007
Acetaminophen hepatotoxicity: A mitochondrial perspective
Abstract
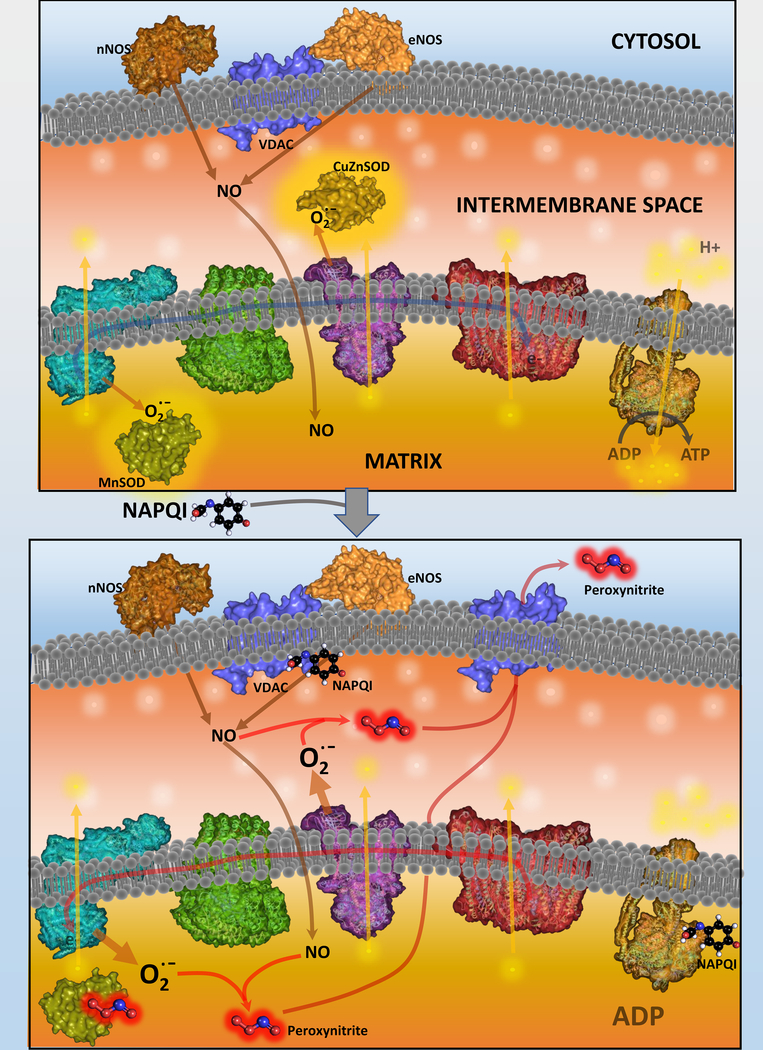
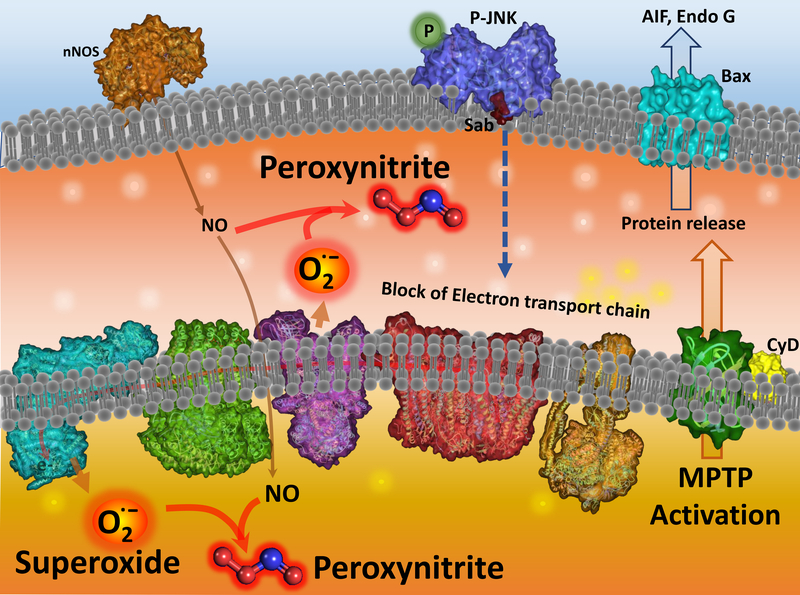
Acetaminophen (APAP) is a highly effective analgesic, which is safe at therapeutic doses. However, an overdose can cause hepatotoxicity and even liver failure. APAP toxicity is currently the most common cause of acute liver failure in the United States. Decades of research on mechanisms of liver injury have established the role of mitochondria as central players in APAP-induced hepatocyte necrosis and this chapter examines the various facets of the organelle's involvement in the process of injury as well as in resolution of damage. The injury process is initiated by formation of a reactive metabolite, which binds to sulfhydryl groups of cellular proteins including mitochondrial proteins. This inhibits the electron transport chain and leads to formation of reactive oxygen species, which induce the activation of redox-sensitive members of the MAP kinase family ultimately causing activation of c-Jun N terminal kinase, JNK. Translocation of JNK to the mitochondria then amplifies mitochondrial dysfunction, ultimately resulting in mitochondrial permeability transition and release of mitochondrial intermembrane proteins, which trigger nuclear DNA fragmentation. Together, these events result in hepatocyte necrosis, while adaptive mechanisms such as mitophagy remove damaged mitochondria and minimize the extent of the injury. This oscillation between recovery and necrosis is predominant in cells at the edge of the necrotic area in the liver, where induction of mitochondrial biogenesis is important for liver regeneration. All these aspects of mitochondria in APAP hepatotoxicity, as well as their relevance to humans with APAP overdose and development of therapeutic approaches will be examined in detail in this chapter.
Keywords: Drug toxicity; Hepatocyte; Liver; Necrosis; Nitric oxide; Superoxide.
© 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Similar articles
-
Mitochondrial protein adduct and superoxide generation are prerequisites for early activation of c-jun N-terminal kinase within the cytosol after an acetaminophen overdose in mice.Toxicol Lett. 2021 Mar 1;338:21-31. doi: 10.1016/j.toxlet.2020.12.005. Epub 2020 Dec 5. Toxicol Lett. 2021. PMID: 33290831 Free PMC article.
-
A mitochondrial journey through acetaminophen hepatotoxicity.Food Chem Toxicol. 2020 Jun;140:111282. doi: 10.1016/j.fct.2020.111282. Epub 2020 Mar 21. Food Chem Toxicol. 2020. PMID: 32209353 Free PMC article. Review.
-
Gab1 adaptor protein acts as a gatekeeper to balance hepatocyte death and proliferation during acetaminophen-induced liver injury in mice.Hepatology. 2016 Apr;63(4):1340-55. doi: 10.1002/hep.28410. Epub 2016 Jan 22. Hepatology. 2016. PMID: 26680679
-
Central Mechanisms of Acetaminophen Hepatotoxicity: Mitochondrial Dysfunction by Protein Adducts and Oxidant Stress.Drug Metab Dispos. 2024 Jul 16;52(8):712-721. doi: 10.1124/dmd.123.001279. Drug Metab Dispos. 2024. PMID: 37567742 Review.
-
Low Dose Acetaminophen Induces Reversible Mitochondrial Dysfunction Associated with Transient c-Jun N-Terminal Kinase Activation in Mouse Liver.Toxicol Sci. 2016 Mar;150(1):204-15. doi: 10.1093/toxsci/kfv319. Epub 2015 Dec 31. Toxicol Sci. 2016. PMID: 26721299 Free PMC article.
Cited by
-
Mitochondria in Acetaminophen-Induced Liver Injury and Recovery: A Concise Review.Livers. 2023 Jun;3(2):219-231. doi: 10.3390/livers3020014. Epub 2023 Apr 10. Livers. 2023. PMID: 37377765 Free PMC article.
-
Neutrophil extracellular traps promote acetaminophen-induced acute liver injury in mice via AIM2.Acta Pharmacol Sin. 2024 Aug;45(8):1660-1672. doi: 10.1038/s41401-024-01239-2. Epub 2024 Apr 8. Acta Pharmacol Sin. 2024. PMID: 38589685
-
Targeting Autophagy for Acetaminophen-Induced Liver Injury: An Update.Livers. 2024 Sep;4(3):377-387. doi: 10.3390/livers4030027. Epub 2024 Aug 14. Livers. 2024. PMID: 39301093 Free PMC article.
-
Biomarkers of drug-induced liver injury: a mechanistic perspective through acetaminophen hepatotoxicity.Expert Rev Gastroenterol Hepatol. 2021 Apr;15(4):363-375. doi: 10.1080/17474124.2021.1857238. Epub 2020 Dec 9. Expert Rev Gastroenterol Hepatol. 2021. PMID: 33242385 Free PMC article. Review.
-
Translocation of Adenosine A2B Receptor to Mitochondria Influences Cytochrome P450 2E1 Activity after Acetaminophen Overdose.Livers. 2024 Mar;4(1):15-30. doi: 10.3390/livers4010002. Epub 2023 Dec 26. Livers. 2024. PMID: 39007013 Free PMC article.
References
-
- Andersson BS, Rundgren M, Nelson SD, & Harder S (1990). N-acetyl-p-benzoquinone imine-induced changes in the energy metabolism in hepatocytes. Chem Biol Interact, 75(2), 201–211. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
