

High-Throughput Sequencing Assists Studies in Genomic Variability and Epidemiology of Little Cherry Virus 1 and 2 infecting Prunus spp. in Belgium
- PMID: 31261922
- PMCID: PMC6669712
- DOI: 10.3390/v11070592
High-Throughput Sequencing Assists Studies in Genomic Variability and Epidemiology of Little Cherry Virus 1 and 2 infecting Prunus spp. in Belgium
Abstract
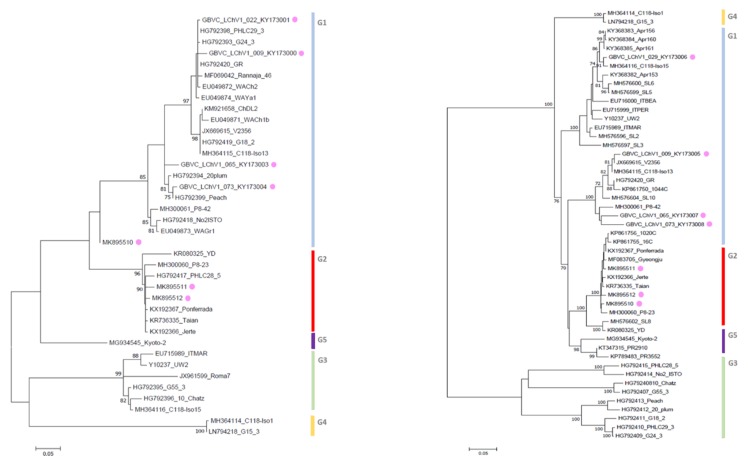
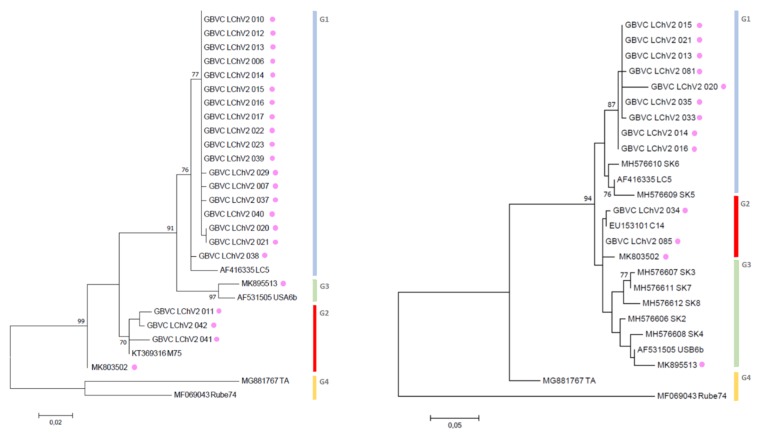
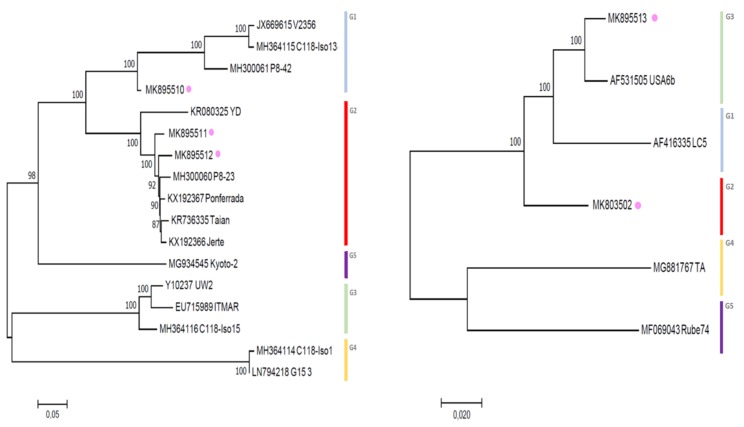
Little cherry disease, caused by little cherry virus 1 (LChV-1) and little cherry virus 2 (LChV-2), which are both members of the family Closteroviridae, severely affects sweet (Prunus avium L.) and sour cherry (P. cerasus L.) orchards lifelong production worldwide. An intensive survey was conducted across different geographic regions of Belgium to study the disease presence on these perennial woody plants and related species. Symptomatic as well as non-symptomatic Prunus spp. trees tested positive via RT-PCR for LChV-1 and -2 in single or mixed infections, with a slightly higher incidence for LChV-1. Both viruses were widespread and highly prevalent in nearly all Prunus production areas as well as in private gardens and urban lane trees. The genetic diversity of Belgian LChV-1 and -2 isolates was assessed by Sanger sequencing of partial genomic regions. A total RNA High-Throughput Sequencing (HTS) approach confirmed the presence of both viruses, and revealed the occurrence of other Prunus-associated viruses, namely cherry virus A (CVA), prune dwarf virus (PDV) and prunus virus F (PrVF). The phylogenetic inference from full-length genomes revealed well-defined evolutionary phylogroups with high genetic variability and diversity for LChV-1 and LChV-2 Belgian isolates, yet with little or no correlation with planting area or cultivated varieties. The global diversity and the prevalence in horticultural areas of LChV-1 and -2 variants, in association with other recently described fruit tree viruses, are of particular concern. Future epidemiological implications as well as new investigation avenues are exhaustively discussed.
Keywords: Ampelovirus; Closteroviridae; HTS; RNA virus phylogeny; Velarivirus; fruit trees and plant viruses; genetic variability; molecular evolution; total RNA.
Conflict of interest statement
The authors declare no conflict of interest. The founding sponsors had no direct role in the design of the study; in the collection, analyses, or interpretation of the data; in the writing of the manuscript, and in the decision to publish the results.
Figures
Similar articles
-
Complete nucleotide sequence of little cherry virus 1 (LChV-1) infecting sweet cherry in China.Arch Virol. 2016 Mar;161(3):749-53. doi: 10.1007/s00705-015-2737-z. Epub 2016 Jan 5. Arch Virol. 2016. PMID: 26733294
-
First Report of Little cherry virus 2 in Flowering and Sweet Cherry Trees in China.Plant Dis. 2011 Nov;95(11):1484. doi: 10.1094/PDIS-10-10-0766. Plant Dis. 2011. PMID: 30731776
-
Genomic detection and characterization of a Korean isolate of Little cherry virus 1 sampled from a peach tree.Virus Genes. 2015 Oct;51(2):260-6. doi: 10.1007/s11262-015-1225-6. Epub 2015 Aug 28. Virus Genes. 2015. PMID: 26315329
-
Ilarviruses of Prunus spp.: a continued concern for fruit trees.Phytopathology. 2012 Dec;102(12):1108-20. doi: 10.1094/PHYTO-02-12-0023-RVW. Phytopathology. 2012. PMID: 23148725 Review.
-
Genomic, Morphological and Biological Traits of the Viruses Infecting Major Fruit Trees.Viruses. 2019 Jun 4;11(6):515. doi: 10.3390/v11060515. Viruses. 2019. PMID: 31167478 Free PMC article. Review.
Cited by
-
Comprehensive Metatranscriptomic Analysis of Plant Viruses in Imported Frozen Cherries and Blueberries.Plant Pathol J. 2024 Aug;40(4):377-389. doi: 10.5423/PPJ.OA.06.2024.0088. Epub 2024 Aug 1. Plant Pathol J. 2024. PMID: 39117336 Free PMC article.
-
First Report and Complete Genome Characterization of Cherry Virus A and Little Cherry Virus 1 from Russia.Plants (Basel). 2023 Sep 18;12(18):3295. doi: 10.3390/plants12183295. Plants (Basel). 2023. PMID: 37765462 Free PMC article.
-
Special Issue "Plant Virus Epidemiology and Control".Viruses. 2020 Mar 12;12(3):309. doi: 10.3390/v12030309. Viruses. 2020. PMID: 32178441 Free PMC article.
-
High-throughput detection of a large set of viruses and viroids of pome and stone fruit trees by multiplex PCR-based amplicon sequencing.Front Plant Sci. 2022 Dec 12;13:1072768. doi: 10.3389/fpls.2022.1072768. eCollection 2022. Front Plant Sci. 2022. PMID: 36578329 Free PMC article.
-
Application of High-Throughput Sequencing for Comprehensive Virome Profiling in Grapevines Shows Yellows in Iran.Viruses. 2024 Jan 29;16(2):204. doi: 10.3390/v16020204. Viruses. 2024. PMID: 38399980 Free PMC article.
References
-
- Katsiani A.T., Maliogka V.I., Amoutzias G.D., Efthimiou K.E., Katis N.I. Insights into the genetic diversity and evolution of Little cherry virus 1. Plant Pathol. 2015;64:817–824. doi: 10.1111/ppa.12309. - DOI
-
- FAO Food and Agriculture organization of the United Nations 2013. [(accessed on 17 January 2019)]; Available online: http://www.fao.org/statistics.
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Miscellaneous
