

Co-SELECT reveals sequence non-specific contribution of DNA shape to transcription factor binding in vitro
- PMID: 31226207
- PMCID: PMC6649817
- DOI: 10.1093/nar/gkz540
Co-SELECT reveals sequence non-specific contribution of DNA shape to transcription factor binding in vitro
Abstract
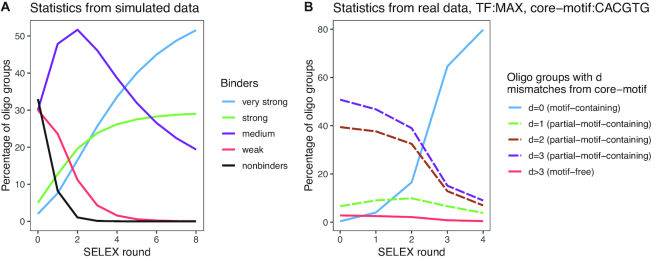
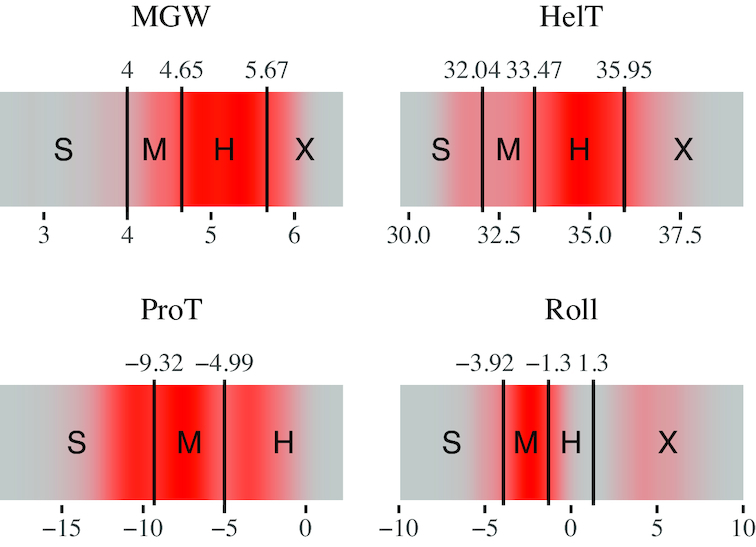
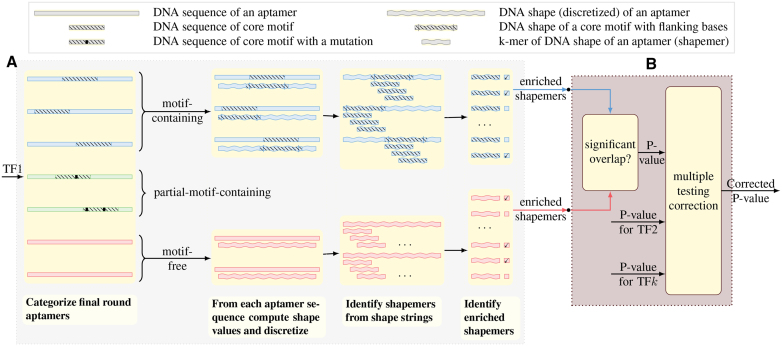
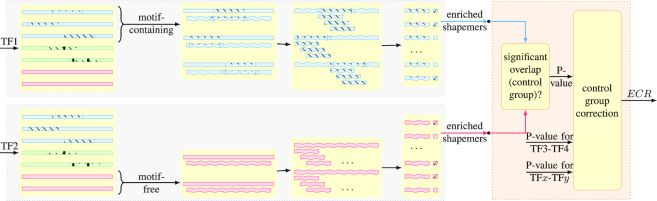
Understanding the principles of DNA binding by transcription factors (TFs) is of primary importance for studying gene regulation. Recently, several lines of evidence suggested that both DNA sequence and shape contribute to TF binding. However, the following compelling question is yet to be considered: in the absence of any sequence similarity to the binding motif, can DNA shape still increase binding probability? To address this challenge, we developed Co-SELECT, a computational approach to analyze the results of in vitro HT-SELEX experiments for TF-DNA binding. Specifically, Co-SELECT leverages the presence of motif-free sequences in late HT-SELEX rounds and their enrichment in weak binders allows Co-SELECT to detect an evidence for the role of DNA shape features in TF binding. Our approach revealed that, even in the absence of the sequence motif, TFs have propensity to bind to DNA molecules of the shape consistent with the motif specific binding. This provides the first direct evidence that shape features that accompany the preferred sequence motifs also bestow an advantage for weak, sequence non-specific binding.
Published by Oxford University Press on behalf of Nucleic Acids Research 2019.
Figures
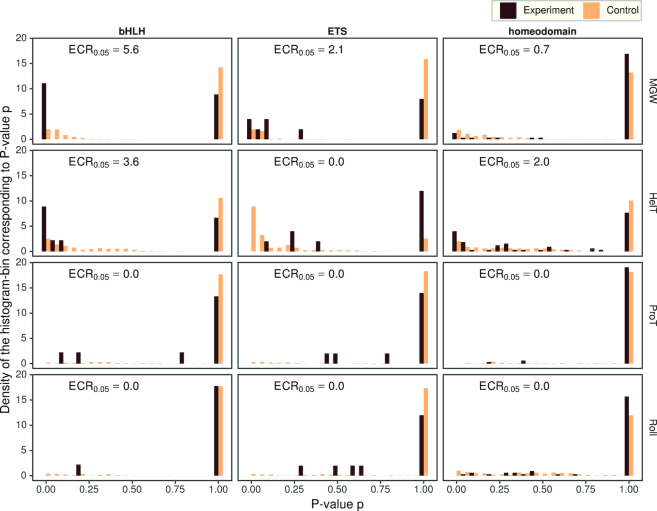
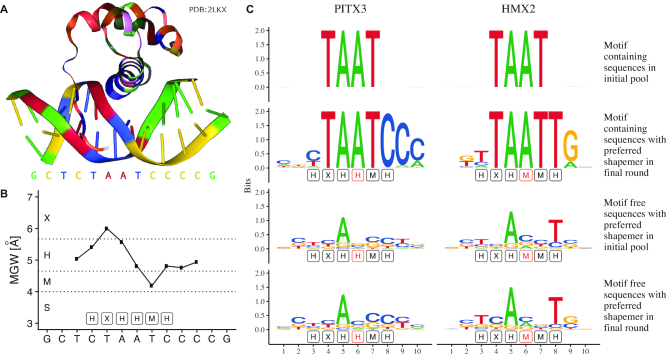
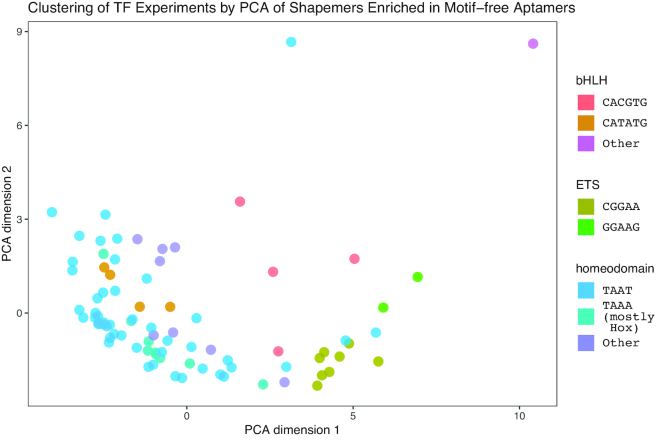
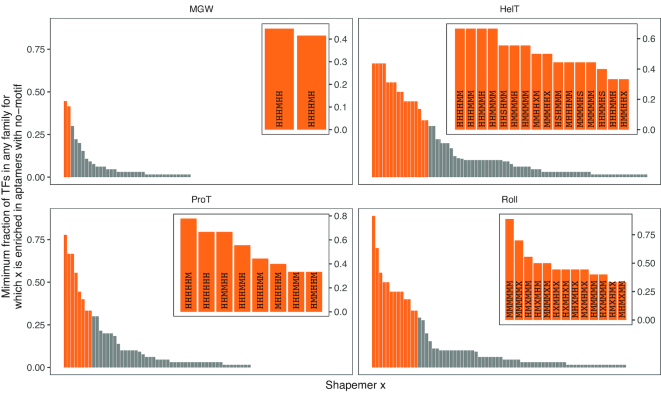
Similar articles
-
RaptRanker: in silico RNA aptamer selection from HT-SELEX experiment based on local sequence and structure information.Nucleic Acids Res. 2020 Aug 20;48(14):e82. doi: 10.1093/nar/gkaa484. Nucleic Acids Res. 2020. PMID: 32537639 Free PMC article.
-
APTANI: a computational tool to select aptamers through sequence-structure motif analysis of HT-SELEX data.Bioinformatics. 2016 Jan 15;32(2):161-4. doi: 10.1093/bioinformatics/btv545. Epub 2015 Sep 22. Bioinformatics. 2016. PMID: 26395772
-
Combining SELEX and the yeast three-hybrid system for in vivo selection and classification of RNA aptamers.RNA. 2007 Apr;13(4):614-22. doi: 10.1261/rna.334307. Epub 2007 Feb 5. RNA. 2007. PMID: 17283213 Free PMC article.
-
Methods developed for SELEX.Anal Bioanal Chem. 2007 Jan;387(1):171-82. doi: 10.1007/s00216-006-0826-2. Epub 2006 Oct 28. Anal Bioanal Chem. 2007. PMID: 17072603 Review.
-
SELEX Modifications and Bioanalytical Techniques for Aptamer-Target Binding Characterization.Crit Rev Anal Chem. 2016 Nov;46(6):521-37. doi: 10.1080/10408347.2016.1157014. Epub 2016 Mar 15. Crit Rev Anal Chem. 2016. PMID: 26980177 Review.
Cited by
-
Noncanonical binding of transcription factors: time to revisit specificity?Mol Biol Cell. 2023 Aug 1;34(9):pe4. doi: 10.1091/mbc.E22-08-0325. Mol Biol Cell. 2023. PMID: 37486893 Free PMC article. Review.
-
TFBSshape: an expanded motif database for DNA shape features of transcription factor binding sites.Nucleic Acids Res. 2020 Jan 8;48(D1):D246-D255. doi: 10.1093/nar/gkz970. Nucleic Acids Res. 2020. PMID: 31665425 Free PMC article.
-
Prediction of regulatory motifs from human Chip-sequencing data using a deep learning framework.Nucleic Acids Res. 2019 Sep 5;47(15):7809-7824. doi: 10.1093/nar/gkz672. Nucleic Acids Res. 2019. PMID: 31372637 Free PMC article.
-
A De Novo Shape Motif Discovery Algorithm Reveals Preferences of Transcription Factors for DNA Shape Beyond Sequence Motifs.Cell Syst. 2019 Jan 23;8(1):27-42.e6. doi: 10.1016/j.cels.2018.12.001. Epub 2019 Jan 16. Cell Syst. 2019. PMID: 30660610 Free PMC article.
-
Bioinformatics pipeline using JUDI: Just Do It!Bioinformatics. 2020 Apr 15;36(8):2572-2574. doi: 10.1093/bioinformatics/btz956. Bioinformatics. 2020. PMID: 31882996 Free PMC article.
References
-
- Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000; 16:16–23. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
