

Analysis of Paramyxovirus Transcription and Replication by High-Throughput Sequencing
- PMID: 31189700
- PMCID: PMC6694822
- DOI: 10.1128/JVI.00571-19
Analysis of Paramyxovirus Transcription and Replication by High-Throughput Sequencing
Abstract
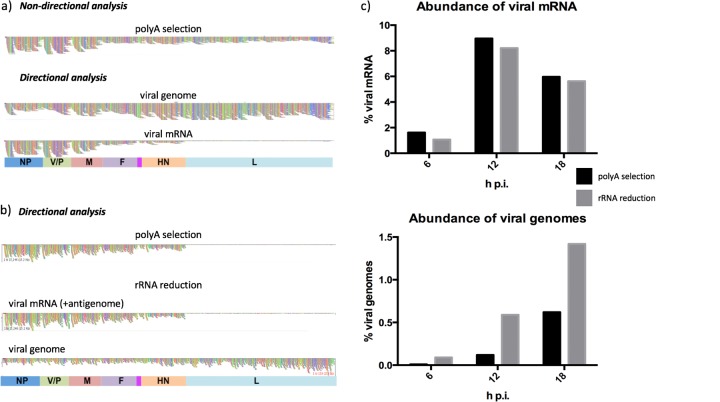
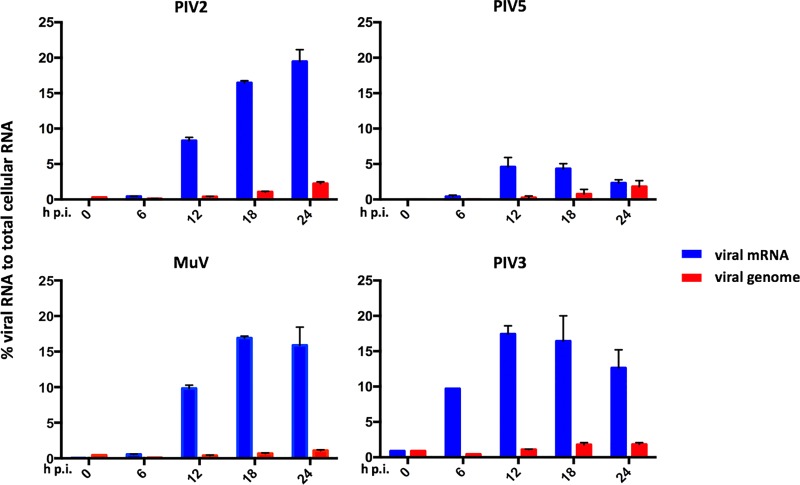
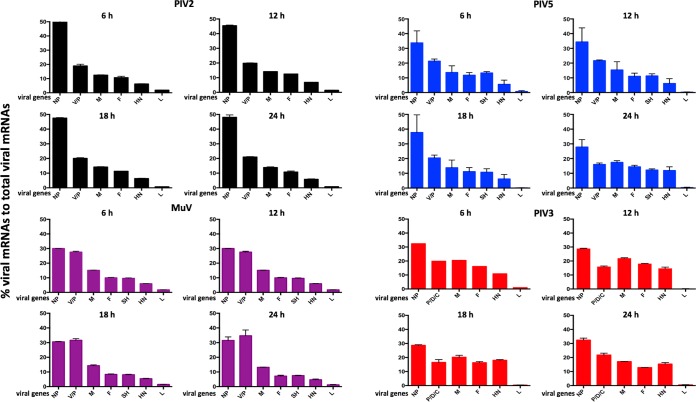
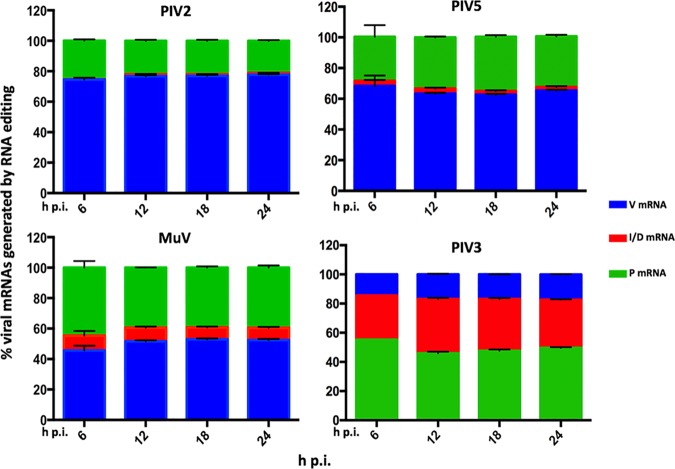
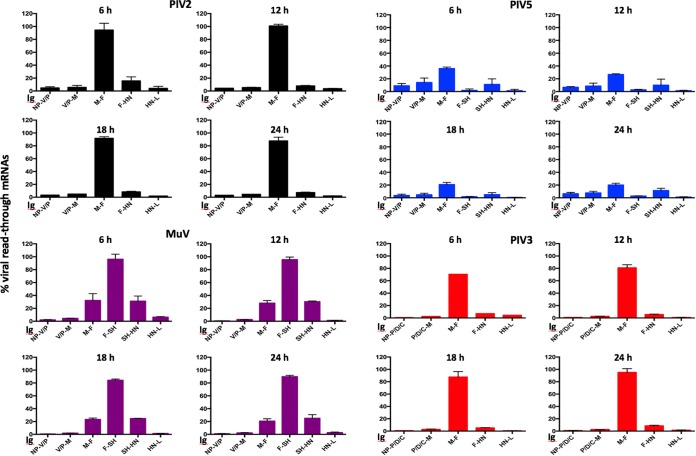
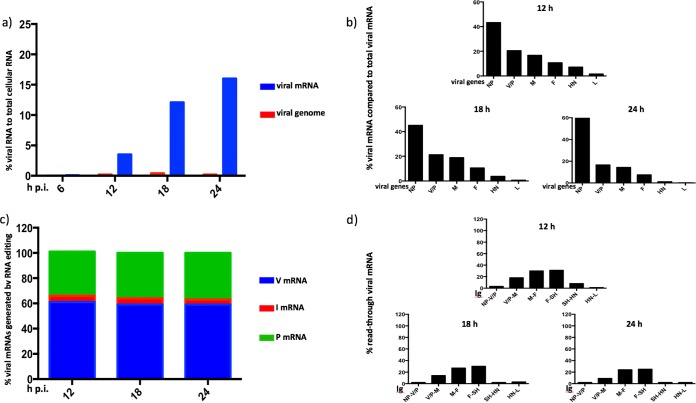
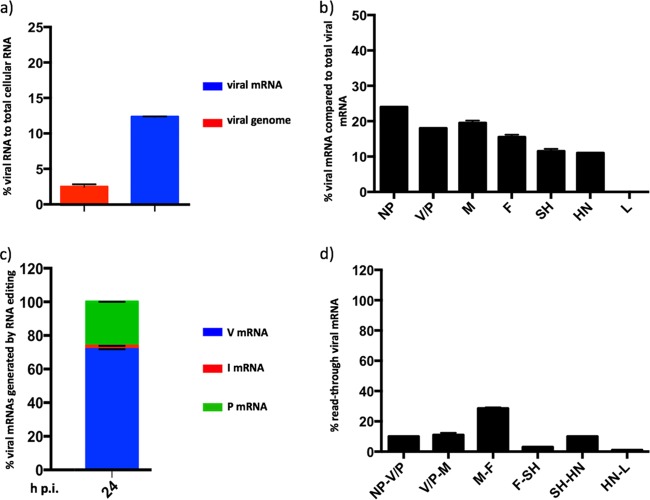
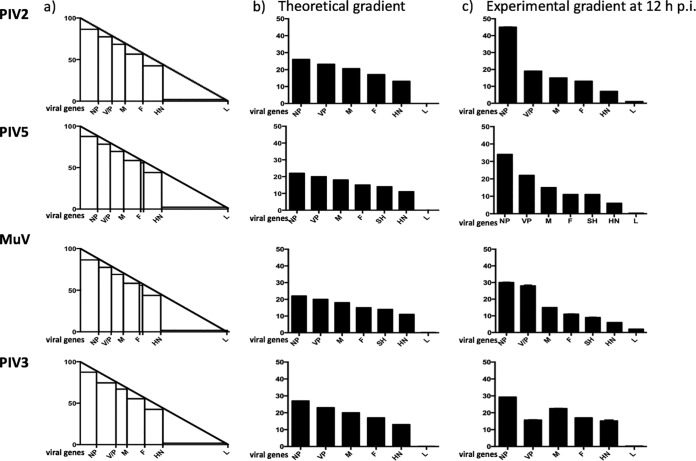
We have developed a high-throughput sequencing (HTS) workflow for investigating paramyxovirus transcription and replication. We show that sequencing of oligo(dT)-selected polyadenylated mRNAs, without considering the orientation of the RNAs from which they had been generated, cannot accurately be used to analyze the abundance of viral mRNAs because genomic RNA copurifies with the viral mRNAs. The best method is directional sequencing of infected cell RNA that has physically been depleted of ribosomal and mitochondrial RNA followed by bioinformatic steps to differentiate data originating from genomes from viral mRNAs and antigenomes. This approach has the advantage that the abundance of viral mRNA (and antigenomes) and genomes can be analyzed and quantified from the same data. We investigated the kinetics of viral transcription and replication during infection of A549 cells with parainfluenza virus type 2 (PIV2), PIV3, PIV5, or mumps virus and determined the abundances of individual viral mRNAs and readthrough mRNAs. We found that the mRNA abundance gradients differed significantly between all four viruses but that for each virus the pattern remained relatively stable throughout infection. We suggest that rapid degradation of non-poly(A) mRNAs may be primarily responsible for the shape of the mRNA abundance gradient in parainfluenza virus 3, whereas a combination of this factor and disengagement of RNA polymerase at intergenic sequences, particularly those at the NP:P and P:M gene boundaries, may be responsible in the other viruses.IMPORTANCE High-throughput sequencing (HTS) of virus-infected cells can be used to study in great detail the patterns of virus transcription and replication. For paramyxoviruses, and by analogy for all other negative-strand RNA viruses, we show that directional sequencing must be used to distinguish between genomic RNA and mRNA/antigenomic RNA because significant amounts of genomic RNA copurify with poly(A)-selected mRNA. We found that the best method is directional sequencing of total cell RNA, after the physical removal of rRNA (and mitochondrial RNA), because quantitative information on the abundance of both genomic RNA and mRNA/antigenomes can be simultaneously derived. Using this approach, we revealed new details of the kinetics of virus transcription and replication for parainfluenza virus (PIV) type 2, PIV3, PIV5, and mumps virus, as well as on the relative abundance of the individual viral mRNAs.
Keywords: high-throughput sequencing; paramyxovirus; replication; transcription.
Copyright © 2019 Wignall-Fleming et al.
Figures
Similar articles
-
Human IFIT1 Inhibits mRNA Translation of Rubulaviruses but Not Other Members of the Paramyxoviridae Family.J Virol. 2016 Sep 29;90(20):9446-56. doi: 10.1128/JVI.01056-16. Print 2016 Oct 15. J Virol. 2016. PMID: 27512068 Free PMC article.
-
Visualization of Arenavirus RNA Species in Individual Cells by Single-Molecule Fluorescence In Situ Hybridization Suggests a Model of Cyclical Infection and Clearance during Persistence.J Virol. 2018 May 29;92(12):e02241-17. doi: 10.1128/JVI.02241-17. Print 2018 Jun 15. J Virol. 2018. PMID: 29643234 Free PMC article.
-
The Nucleocapsid of Paramyxoviruses: Structure and Function of an Encapsidated Template.Viruses. 2021 Dec 9;13(12):2465. doi: 10.3390/v13122465. Viruses. 2021. PMID: 34960734 Free PMC article. Review.
-
Structure of the Paramyxovirus Parainfluenza Virus 5 Nucleoprotein in Complex with an Amino-Terminal Peptide of the Phosphoprotein.J Virol. 2018 Feb 12;92(5):e01304-17. doi: 10.1128/JVI.01304-17. Print 2018 Mar 1. J Virol. 2018. PMID: 29237836 Free PMC article.
-
Initiation and regulation of paramyxovirus transcription and replication.Virology. 2015 May;479-480:545-54. doi: 10.1016/j.virol.2015.01.014. Epub 2015 Feb 13. Virology. 2015. PMID: 25683441 Free PMC article. Review.
Cited by
-
Human Cytomegalovirus Long Non-coding RNA1.2 Suppresses Extracellular Release of the Pro-inflammatory Cytokine IL-6 by Blocking NF-κB Activation.Front Cell Infect Microbiol. 2020 Jul 22;10:361. doi: 10.3389/fcimb.2020.00361. eCollection 2020. Front Cell Infect Microbiol. 2020. PMID: 32793512 Free PMC article.
-
Single-Cell Transcriptome Atlas of Newcastle Disease Virus in Chickens Both In Vitro and In Vivo.Microbiol Spectr. 2023 Jun 15;11(3):e0512122. doi: 10.1128/spectrum.05121-22. Epub 2023 May 16. Microbiol Spectr. 2023. PMID: 37191506 Free PMC article.
-
Re-assessing the diversity of negative strand RNA viruses in insects.PLoS Pathog. 2019 Dec 12;15(12):e1008224. doi: 10.1371/journal.ppat.1008224. eCollection 2019 Dec. PLoS Pathog. 2019. PMID: 31830128 Free PMC article.
-
Engineering of Live Chimeric Vaccines against Human Metapneumovirus.Pathogens. 2020 Feb 19;9(2):135. doi: 10.3390/pathogens9020135. Pathogens. 2020. PMID: 32093057 Free PMC article. Review.
-
Patterns of RNA Editing in Newcastle Disease Virus Infections.Viruses. 2020 Nov 2;12(11):1249. doi: 10.3390/v12111249. Viruses. 2020. PMID: 33147786 Free PMC article.
References
-
- Samal SK. 2011. The biology of paramyxoviruses. Caister Academic Press, Wymondham, England.
-
- Lamb R. 2013. Paramyxoviridae: the viruses and their replication, 6th ed Lippincott, Williams and Wilkins, Philadelphia, PA.
-
- Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol 283:61–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
