

Induction of anti-cancer T cell immunity by in situ vaccination using systemically administered nanomedicines
- PMID: 31185250
- PMCID: PMC6629511
- DOI: 10.1016/j.canlet.2019.114427
Induction of anti-cancer T cell immunity by in situ vaccination using systemically administered nanomedicines
Abstract
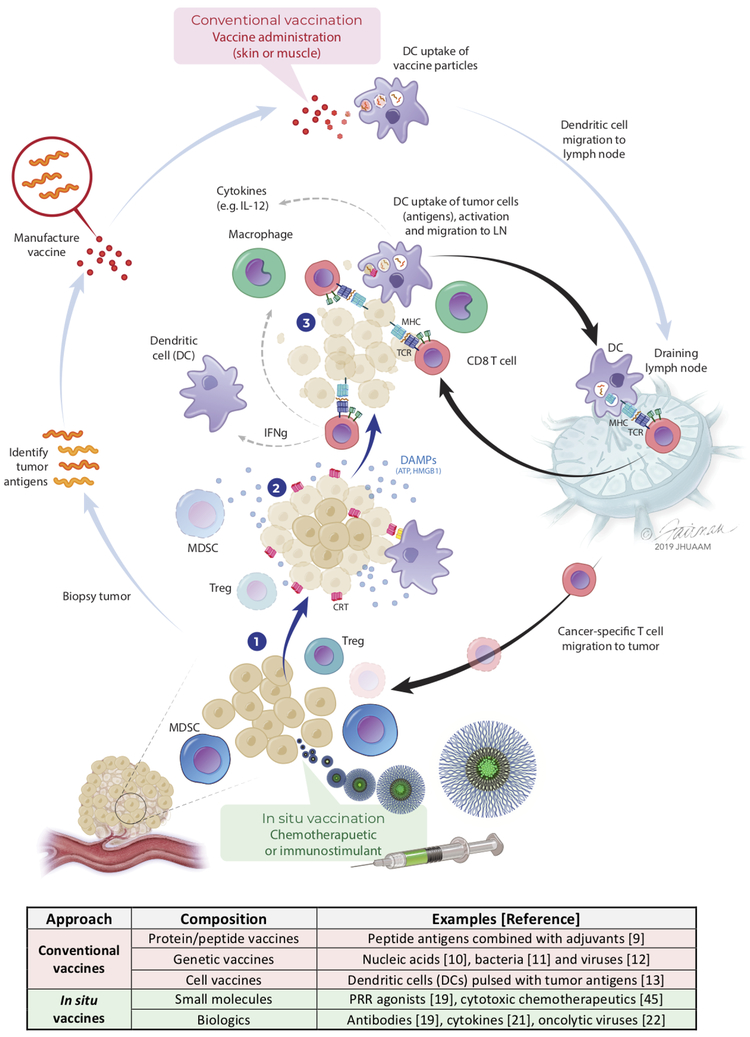
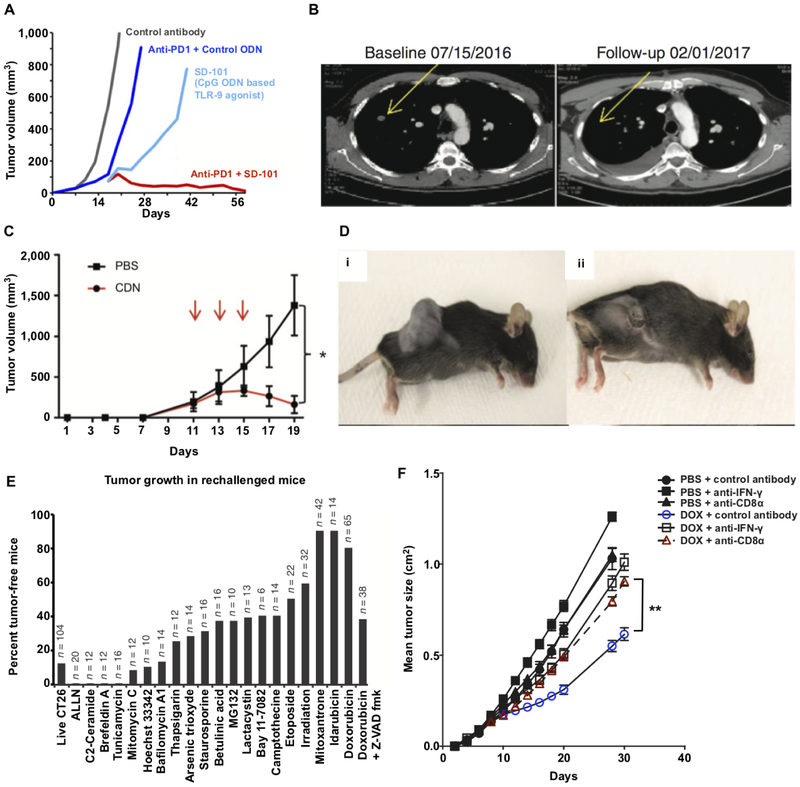
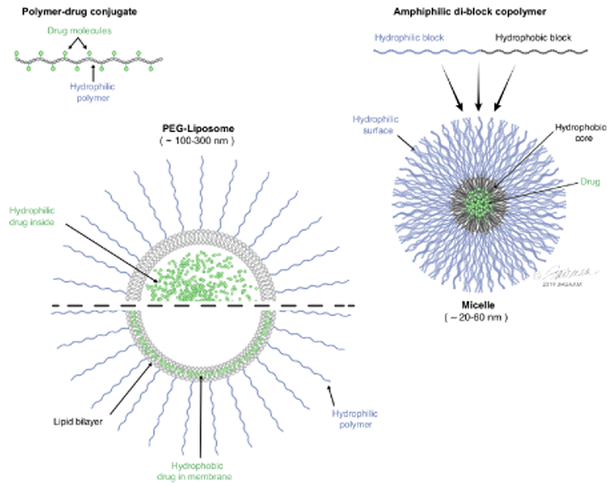
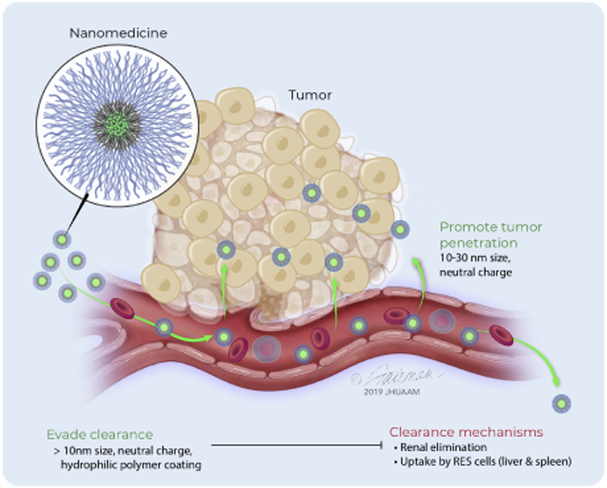
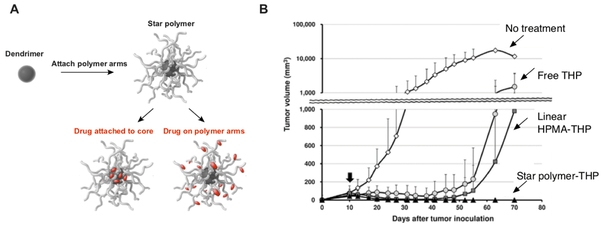
Patients with inadequate anti-cancer T cell responses experience limited benefit from immune checkpoint inhibitors and other immunotherapies that require T cells. Therefore, treatments that induce de novo anti-cancer T cell immunity are needed. One strategy - referred to as in situ vaccination - is to deliver chemotherapeutic or immunostimulatory drugs into tumors to promote cancer cell death and provide a stimulatory environment for priming T cells against antigens already present in the tumor. However, achieving sufficient drug concentrations in tumors without causing dose-limiting toxicities remains a major challenge. To address this challenge, nanomedicines based on nano-sized carriers ('nanocarriers') of chemotherapeutics and immunostimulants are being developed to improve drug accumulation in tumors following systemic (intravenous) administration. Herein, we present the rationale for using systemically administrable nanomedicines to induce anti-cancer T cell immunity via in situ vaccination and provide an overview of synthetic nanomedicines currently used clinically. We also describe general strategies for improving nanomedicine design to increase tumor uptake, including use of micelle- and star polymer-based nanocarriers. We conclude with perspectives for how nanomedicine properties, host factors and treatment combinations can be leveraged to maximize efficacy.
Keywords: Chemotherapeutic and immunostimulant; Immunogenic cell death; Nanomedicine and biomaterials; Nanoparticle and microparticle; Pattern recognition receptor.
Published by Elsevier B.V.
Figures
Similar articles
-
Combining Nanomedicine and Immunotherapy.Acc Chem Res. 2019 Jun 18;52(6):1543-1554. doi: 10.1021/acs.accounts.9b00148. Epub 2019 May 23. Acc Chem Res. 2019. PMID: 31120725 Free PMC article.
-
Tumor-Targeted Nanomedicine for Immunotherapy.Acc Chem Res. 2020 Dec 15;53(12):2765-2776. doi: 10.1021/acs.accounts.0c00518. Epub 2020 Nov 8. Acc Chem Res. 2020. PMID: 33161717 Review.
-
Nanomedicines for an Enhanced Immunogenic Cell Death-Based In Situ Cancer Vaccination Response.Acc Chem Res. 2024 Mar 19;57(6):905-918. doi: 10.1021/acs.accounts.3c00771. Epub 2024 Feb 28. Acc Chem Res. 2024. PMID: 38417027
-
Emerging advances in synthetic cancer nano-vaccines: opportunities and challenges.Expert Rev Vaccines. 2020 Nov;19(11):1053-1071. doi: 10.1080/14760584.2020.1858058. Epub 2020 Dec 14. Expert Rev Vaccines. 2020. PMID: 33315512 Review.
-
Engineered Nanoparticles for Cancer Vaccination and Immunotherapy.Acc Chem Res. 2020 Oct 20;53(10):2094-2105. doi: 10.1021/acs.accounts.0c00456. Epub 2020 Oct 5. Acc Chem Res. 2020. PMID: 33017150 Free PMC article.
Cited by
-
Therapeutic cancer vaccines for pediatric malignancies: advances, challenges, and emerging technologies.Neurooncol Adv. 2021 Feb 11;3(1):vdab027. doi: 10.1093/noajnl/vdab027. eCollection 2021 Jan-Dec. Neurooncol Adv. 2021. PMID: 33860227 Free PMC article. Review.
-
New emerging targets in cancer immunotherapy: the role of neoantigens.ESMO Open. 2020 Apr;4(Suppl 3):e000684. doi: 10.1136/esmoopen-2020-000684. ESMO Open. 2020. PMID: 32269031 Free PMC article. Review.
-
Phage display as a tool for vaccine and immunotherapy development.Bioeng Transl Med. 2019 Sep 18;5(1):e10142. doi: 10.1002/btm2.10142. eCollection 2020 Jan. Bioeng Transl Med. 2019. PMID: 31989033 Free PMC article. Review.
-
Harnessing Biomaterials to Study and Direct Antigen-Specific Immunotherapy.ACS Appl Bio Mater. 2023 Jun 19;6(6):2017-2028. doi: 10.1021/acsabm.3c00136. Epub 2023 Apr 17. ACS Appl Bio Mater. 2023. PMID: 37068126 Free PMC article. Review.
-
Bacteria as Nanoparticle Carriers for Immunotherapy in Oncology.Pharmaceutics. 2022 Apr 3;14(4):784. doi: 10.3390/pharmaceutics14040784. Pharmaceutics. 2022. PMID: 35456618 Free PMC article. Review.
References
-
- Schumacher TN, Schreiber RD, Neoantigens in cancer immunotherapy, Science, 348 (2015) 69–74. - PubMed
-
- Allison JP, Immune Checkpoint Blockade in Cancer Therapy: The 2015 Lasker-DeBakey Clinical Medical Research Award, JAMA, 314 (2015) 1113–1114. - PubMed
-
- Chen DS, Mellman I, Oncology meets immunology: the cancer-immunity cycle, Immunity, 39 (2013) 1–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
