

Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes
- PMID: 31155496
- PMCID: PMC6610720
- DOI: 10.1016/j.cmet.2019.05.006
Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes
Abstract
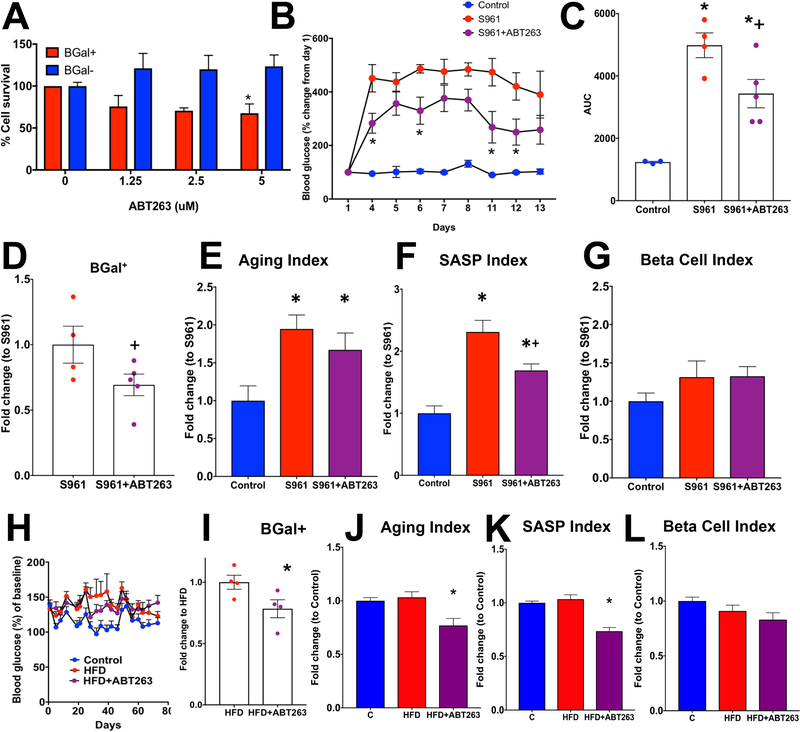
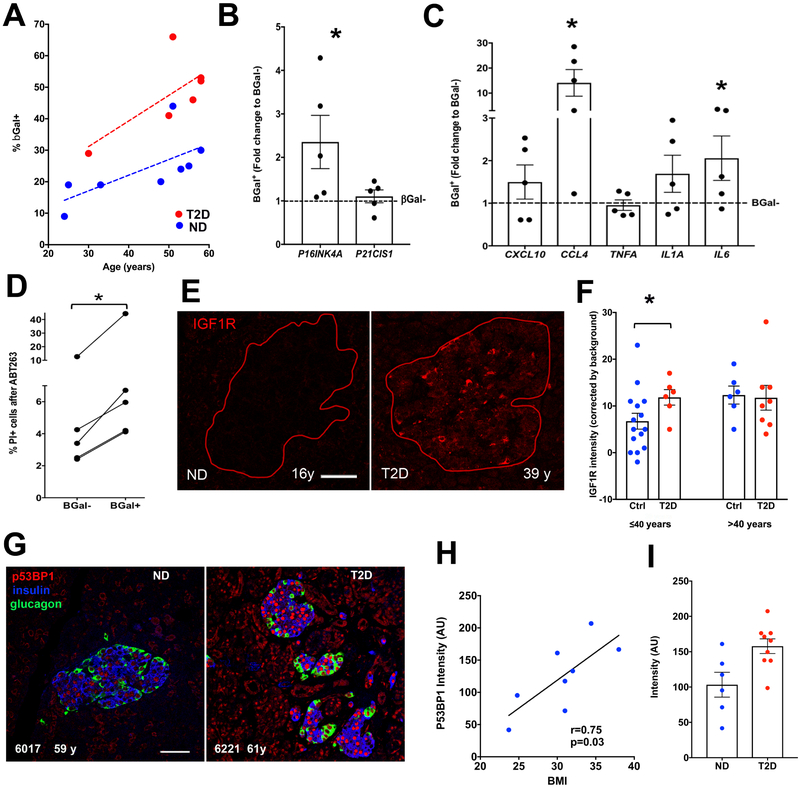
Type 2 diabetes (T2D) is an age-related disease. Although changes in function and proliferation of aged β cells resemble those preceding the development of diabetes, the contribution of β cell aging and senescence remains unclear. We generated a β cell senescence signature and found that insulin resistance accelerates β cell senescence leading to loss of function and cellular identity and worsening metabolic profile. Senolysis (removal of senescent cells), using either a transgenic INK-ATTAC model or oral ABT263, improved glucose metabolism and β cell function while decreasing expression of markers of aging, senescence, and senescence-associated secretory profile (SASP). Beneficial effects of senolysis were observed in an aging model as well as with insulin resistance induced both pharmacologically (S961) and physiologically (high-fat diet). Human senescent β cells also responded to senolysis, establishing the foundation for translation. These novel findings lay the framework to pursue senolysis of β cells as a preventive and alleviating strategy for T2D.
Keywords: SASP; beta cells; glucose metabolism; insulin resistance; insulin secretion; senescence; senescence signature; senescence-associated secretory profile; senolytic therapies; type 2 diabetes.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures





Comment in
-
Targeting β-cell senescence.Nat Rev Endocrinol. 2019 Aug;15(8):438-439. doi: 10.1038/s41574-019-0232-4. Nat Rev Endocrinol. 2019. PMID: 31239521 No abstract available.
Similar articles
-
Cellular Senescence in Diabetes Mellitus: Distinct Senotherapeutic Strategies for Adipose Tissue and Pancreatic β Cells.Front Endocrinol (Lausanne). 2022 Mar 31;13:869414. doi: 10.3389/fendo.2022.869414. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35432205 Free PMC article. Review.
-
Clearance of p16Ink4a-positive cells in a mouse transgenic model does not change β-cell mass and has limited effects on their proliferative capacity.Aging (Albany NY). 2023 Jan 12;15(2):441-458. doi: 10.18632/aging.204483. Epub 2023 Jan 12. Aging (Albany NY). 2023. PMID: 36640267 Free PMC article.
-
Unique Human and Mouse β-Cell Senescence-Associated Secretory Phenotype (SASP) Reveal Conserved Signaling Pathways and Heterogeneous Factors.Diabetes. 2021 May;70(5):1098-1116. doi: 10.2337/db20-0553. Epub 2021 Mar 5. Diabetes. 2021. PMID: 33674410 Free PMC article.
-
β-cell senescence in type 2 diabetes.Aging (Albany NY). 2019 Nov 25;11(22):9967-9968. doi: 10.18632/aging.102502. Epub 2019 Nov 25. Aging (Albany NY). 2019. PMID: 31767811 Free PMC article. No abstract available.
-
Functional changes in beta cells during ageing and senescence.Diabetologia. 2020 Oct;63(10):2022-2029. doi: 10.1007/s00125-020-05185-6. Epub 2020 Sep 7. Diabetologia. 2020. PMID: 32894312 Free PMC article. Review.
Cited by
-
Cell senescence in cardiometabolic diseases.NPJ Aging. 2024 Oct 21;10(1):46. doi: 10.1038/s41514-024-00170-4. NPJ Aging. 2024. PMID: 39433786 Free PMC article. Review.
-
Calorie restriction increases insulin sensitivity to promote beta cell homeostasis and longevity in mice.Nat Commun. 2024 Oct 21;15(1):9063. doi: 10.1038/s41467-024-53127-2. Nat Commun. 2024. PMID: 39433757 Free PMC article.
-
Inflammatory Trajectory of Type 2 Diabetes: Novel Opportunities for Early and Late Treatment.Cells. 2024 Oct 8;13(19):1662. doi: 10.3390/cells13191662. Cells. 2024. PMID: 39404426 Free PMC article. Review.
-
Accelerated biological aging, mediating amino acids, and risk of incident type 2 diabetes: a prospective cohort study.J Endocrinol Invest. 2024 Oct 3. doi: 10.1007/s40618-024-02436-w. Online ahead of print. J Endocrinol Invest. 2024. PMID: 39361235
-
Type 2 diabetes mellitus in adults: pathogenesis, prevention and therapy.Signal Transduct Target Ther. 2024 Oct 2;9(1):262. doi: 10.1038/s41392-024-01951-9. Signal Transduct Target Ther. 2024. PMID: 39353925 Free PMC article. Review.
References
-
- Bliss CR, and Sharp GW (1992). Glucose-induced insulin release in islets of young rats: time-dependent potentiation and effects of 2-bromostearate. Am J Physiol 263, E890–896. - PubMed
-
- Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, and Kahn CR (1997). Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 88, 561–572. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
