

Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy
- PMID: 31126310
- PMCID: PMC6533683
- DOI: 10.1186/s12943-019-1030-2
Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy
Abstract
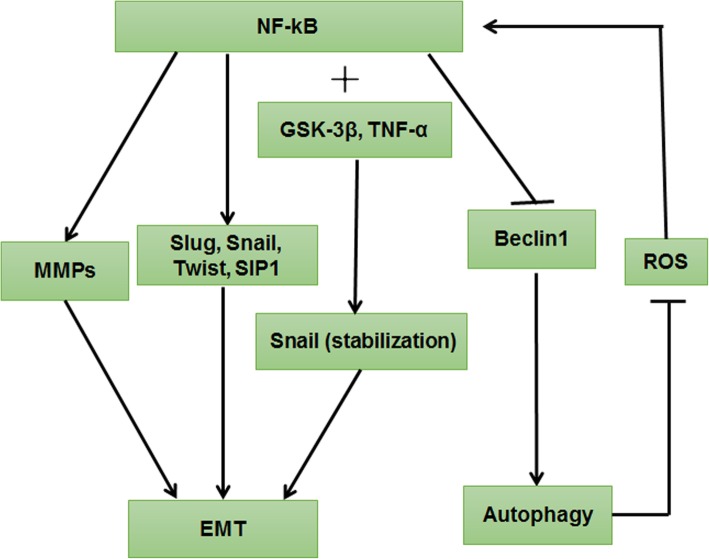
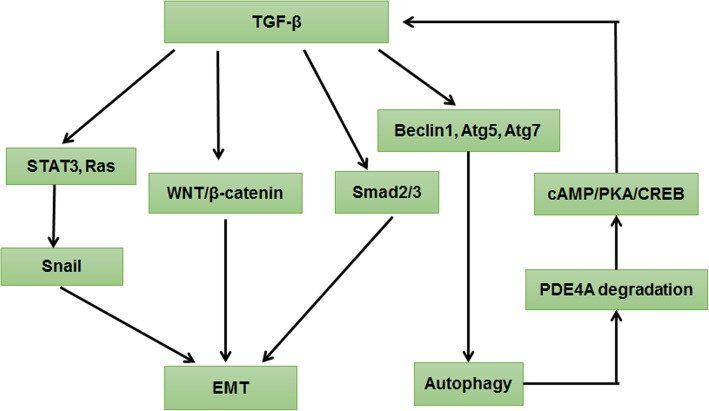
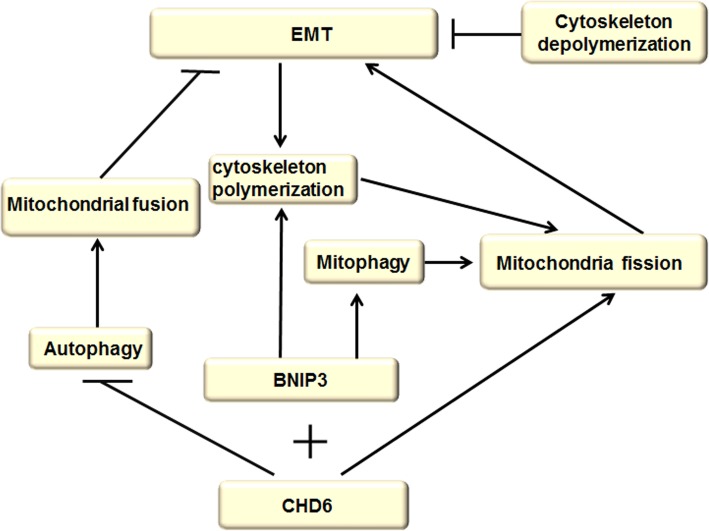
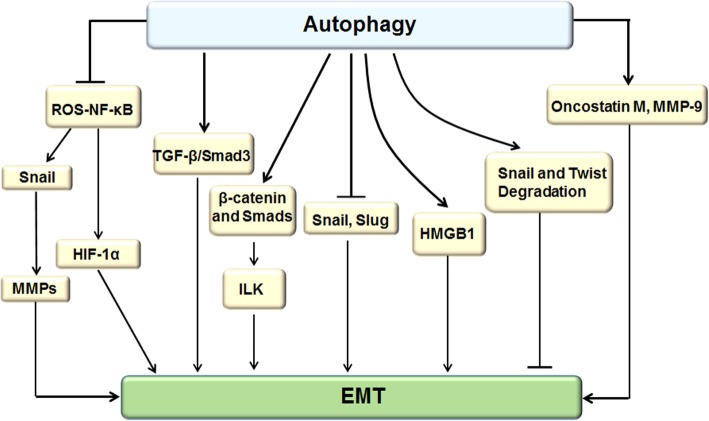
Autophagy is a highly conserved catabolic process that mediates degradation of pernicious or dysfunctional cellular components, such as invasive pathogens, senescent proteins, and organelles. It can promote or suppress tumor development, so it is a "double-edged sword" in tumors that depends on the cell and tissue types and the stages of tumor. The epithelial-mesenchymal transition (EMT) is a complex biological trans-differentiation process that allows epithelial cells to transiently obtain mesenchymal features, including motility and metastatic potential. EMT is considered as an important contributor to the invasion and metastasis of cancers. Thus, clarifying the crosstalk between autophagy and EMT will provide novel targets for cancer therapy. It was reported that EMT-related signal pathways have an impact on autophagy; conversely, autophagy activation can suppress or strengthen EMT by regulating various signaling pathways. On one hand, autophagy activation provides energy and basic nutrients for EMT during metastatic spreading, which assists cells to survive in stressful environmental and intracellular conditions. On the other hand, autophagy, acting as a cancer-suppressive function, is inclined to hinder metastasis by selectively down-regulating critical transcription factors of EMT in the early phases. Therefore, the inhibition of EMT by autophagy inhibitors or activators might be a novel strategy that provides thought and enlightenment for the treatment of cancer. In this article, we discuss in detail the role of autophagy and EMT in the development of cancers, the regulatory mechanisms between autophagy and EMT, the effects of autophagy inhibition or activation on EMT, and the potential applications in anticancer therapy.
Keywords: Anticancer therapy; Autophagy; Cancer metastasis; Epithelial-mesenchymal transition.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


Similar articles
-
Cross talk between autophagy and oncogenic signaling pathways and implications for cancer therapy.Biochim Biophys Acta Rev Cancer. 2021 Aug;1876(1):188565. doi: 10.1016/j.bbcan.2021.188565. Epub 2021 May 13. Biochim Biophys Acta Rev Cancer. 2021. PMID: 33992723 Review.
-
Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer.Cell Death Dis. 2016 Dec 8;7(12):e2520. doi: 10.1038/cddis.2016.415. Cell Death Dis. 2016. PMID: 27929542 Free PMC article. Review.
-
Narrower insight to SIRT1 role in cancer: A potential therapeutic target to control epithelial-mesenchymal transition in cancer cells.J Cell Physiol. 2018 Jun;233(6):4443-4457. doi: 10.1002/jcp.26302. Epub 2018 Jan 15. J Cell Physiol. 2018. PMID: 29194618 Review.
-
Roles of Dietary Phytoestrogens on the Regulation of Epithelial-Mesenchymal Transition in Diverse Cancer Metastasis.Toxins (Basel). 2016 May 24;8(6):162. doi: 10.3390/toxins8060162. Toxins (Basel). 2016. PMID: 27231938 Free PMC article. Review.
-
Alisertib, an Aurora kinase A inhibitor, induces apoptosis and autophagy but inhibits epithelial to mesenchymal transition in human epithelial ovarian cancer cells.Drug Des Devel Ther. 2015 Jan 9;9:425-64. doi: 10.2147/DDDT.S74062. eCollection 2015. Drug Des Devel Ther. 2015. PMID: 25624750 Free PMC article.
Cited by
-
The Roles of ceRNAs-Mediated Autophagy in Cancer Chemoresistance and Metastasis.Cancers (Basel). 2020 Oct 11;12(10):2926. doi: 10.3390/cancers12102926. Cancers (Basel). 2020. PMID: 33050642 Free PMC article. Review.
-
The Potential of Glycyrrhiza from "Medicine Food Homology" in the Fight against Digestive System Tumors.Molecules. 2023 Nov 22;28(23):7719. doi: 10.3390/molecules28237719. Molecules. 2023. PMID: 38067451 Free PMC article. Review.
-
Chemosensitivity enhanced by autophagy inhibition based on a polycationic nano-drug carrier.Nanoscale Adv. 2021 Jan 27;3(6):1656-1673. doi: 10.1039/d0na00990c. eCollection 2021 Mar 23. Nanoscale Adv. 2021. PMID: 36132550 Free PMC article.
-
Interplay of autophagy and cancer stem cells in hepatocellular carcinoma.Mol Biol Rep. 2021 Apr;48(4):3695-3717. doi: 10.1007/s11033-021-06334-9. Epub 2021 Apr 24. Mol Biol Rep. 2021. PMID: 33893928 Review.
-
Cudraxanthone D Regulates Epithelial-Mesenchymal Transition by Autophagy Inhibition in Oral Squamous Cell Carcinoma Cell Lines.Evid Based Complement Alternat Med. 2019 Oct 31;2019:5213028. doi: 10.1155/2019/5213028. eCollection 2019. Evid Based Complement Alternat Med. 2019. PMID: 31781271 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
