

Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells
- PMID: 31121863
- PMCID: PMC6566956
- DOI: 10.3390/ijms20102518
Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells
Abstract
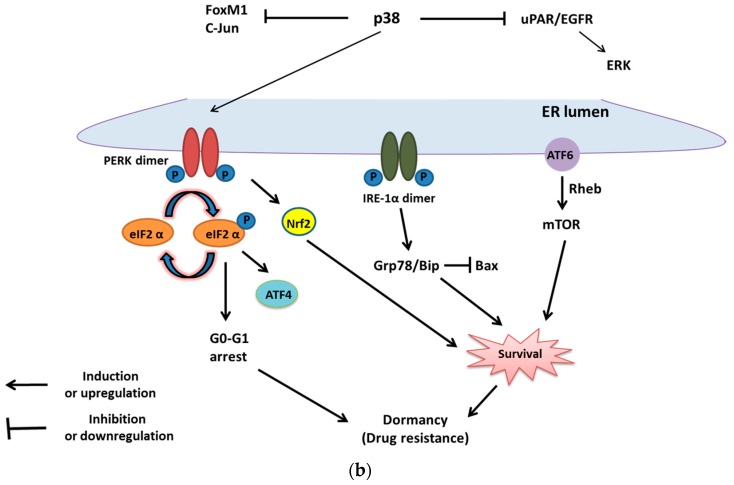
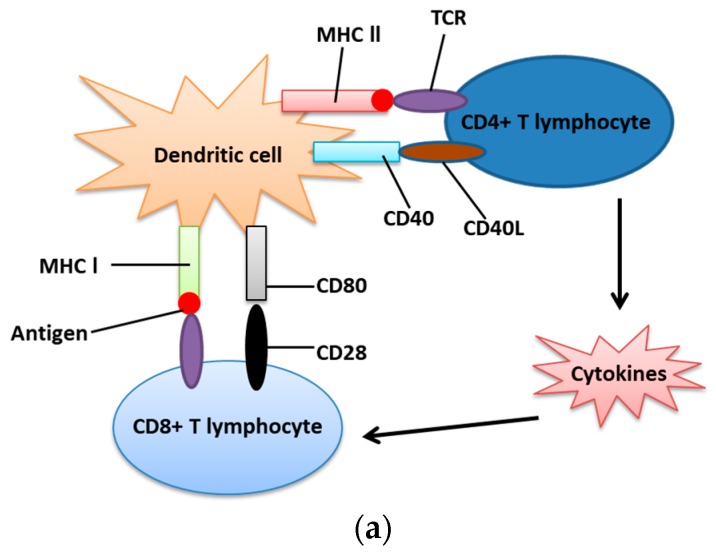
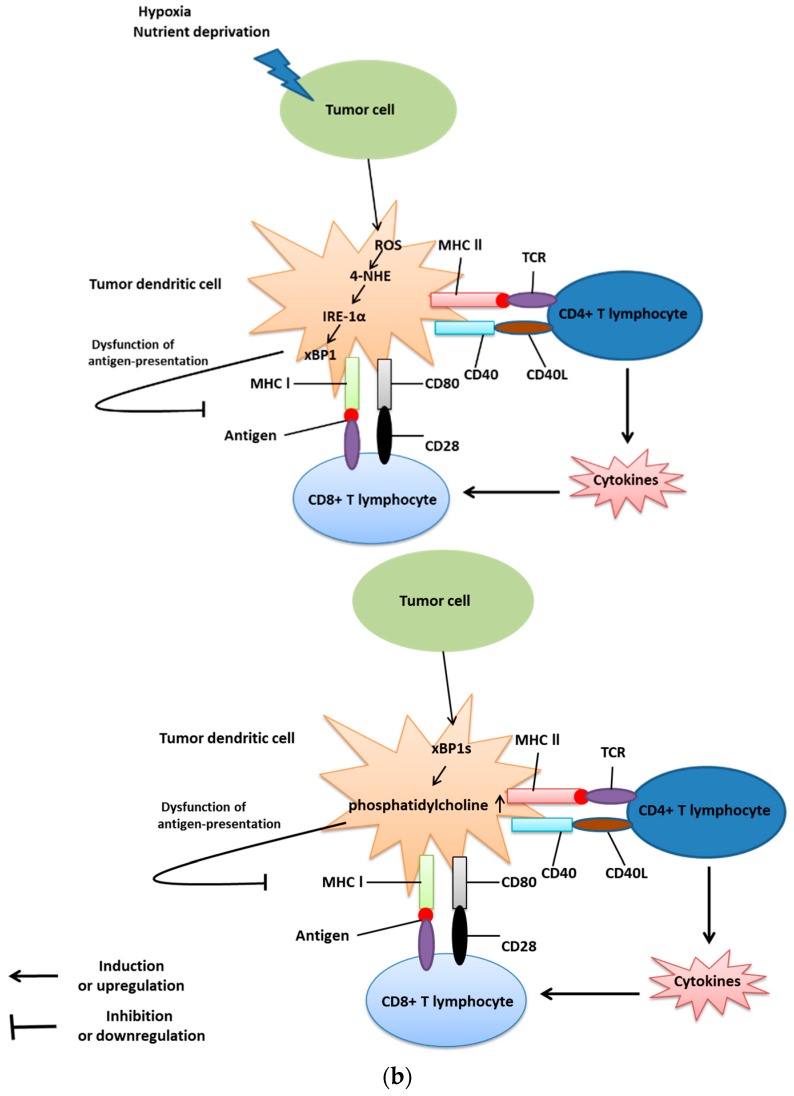
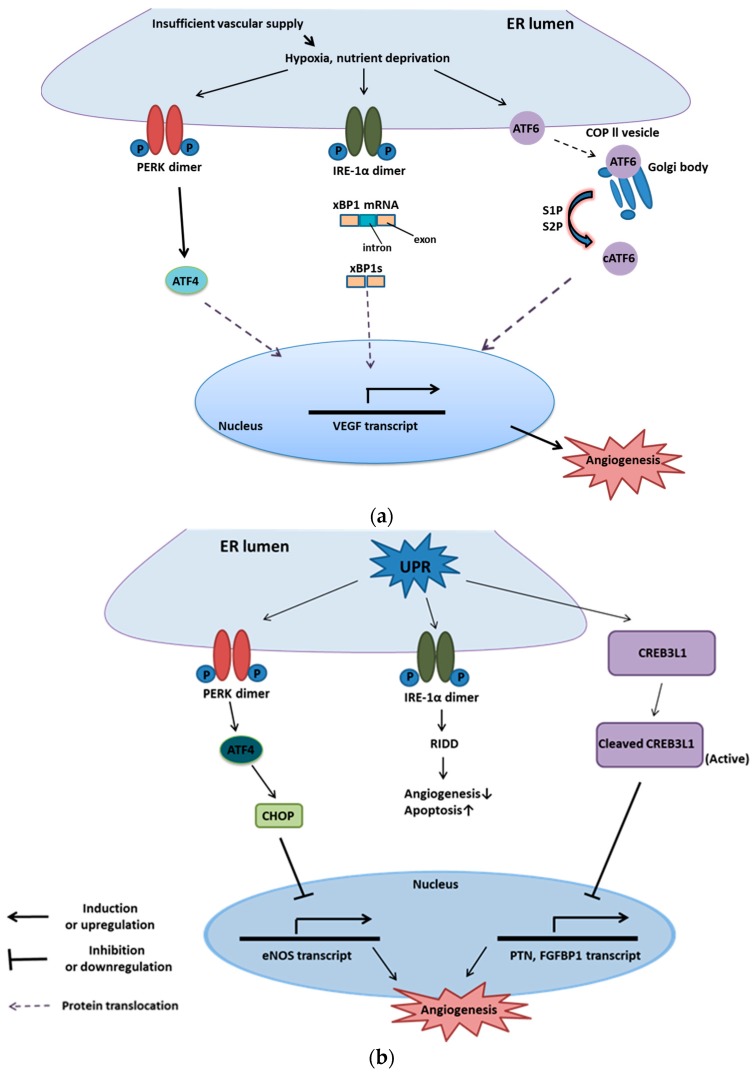
The endoplasmic reticulum (ER) has diverse functions, and especially misfolded protein modification is in the focus of this review paper. With a highly regulatory mechanism, called unfolded protein response (UPR), it protects cells from the accumulation of misfolded proteins. Nevertheless, not only does UPR modify improper proteins, but it also degrades proteins that are unable to recover. Three pathways of UPR, namely PERK, IRE-1, and ATF6, have a significant role in regulating stress-induced physiological responses in cells. The dysregulated UPR may be involved in diseases, such as atherosclerosis, heart diseases, amyotrophic lateral sclerosis (ALS), and cancer. Here, we discuss the relation between UPR and cancer, considering several aspects including survival, dormancy, immunosuppression, angiogenesis, and metastasis of cancer cells. Although several moderate adversities can subject cancer cells to a hostile environment, UPR can ensure their survival. Excessive unfavorable conditions, such as overloading with misfolded proteins and nutrient deprivation, tend to trigger cancer cell death signaling. Regarding dormancy and immunosuppression, cancer cells can survive chemotherapies and acquire drug resistance through dormancy and immunosuppression. Cancer cells can also regulate the downstream of UPR to modulate angiogenesis and promote metastasis. In the end, regulating UPR through different molecular mechanisms may provide promising anticancer treatment options by suppressing cancer proliferation and progression.
Keywords: ATF6; IRE-1; PERK; cancer; endoplasmic reticulum (ER); unfolded protein response (UPR).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
An Overview of Unfolded Protein Response Signaling and Its Role in Cancer.Cancer Biother Radiopharm. 2017 Oct;32(8):275-281. doi: 10.1089/cbr.2017.2309. Cancer Biother Radiopharm. 2017. PMID: 29053418 Review.
-
Driving Cancer Tumorigenesis and Metastasis Through UPR Signaling.Curr Top Microbiol Immunol. 2018;414:159-192. doi: 10.1007/82_2017_36. Curr Top Microbiol Immunol. 2018. PMID: 28710693 Review.
-
Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer.Cell. 2017 Feb 9;168(4):692-706. doi: 10.1016/j.cell.2016.12.004. Cell. 2017. PMID: 28187289 Free PMC article. Review.
-
A Systems Biological View of Life-and-Death Decision with Respect to Endoplasmic Reticulum Stress-The Role of PERK Pathway.Int J Mol Sci. 2017 Jan 5;18(1):58. doi: 10.3390/ijms18010058. Int J Mol Sci. 2017. PMID: 28067773 Free PMC article.
-
The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance.Biol Cell. 2019 Jan;111(1):1-17. doi: 10.1111/boc.201800050. Epub 2018 Oct 29. Biol Cell. 2019. PMID: 30302777 Review.
Cited by
-
Maternal exposure to PM2.5 induces the testicular cell apoptosis in offspring triggered by the UPR-mediated JNK pathway.Toxicol Res (Camb). 2022 Jan 26;11(1):226-234. doi: 10.1093/toxres/tfab116. eCollection 2022 Feb. Toxicol Res (Camb). 2022. PMID: 35237427 Free PMC article.
-
The role of platelets in the regulation of tumor growth and metastasis: the mechanisms and targeted therapy.MedComm (2020). 2023 Sep 14;4(5):e350. doi: 10.1002/mco2.350. eCollection 2023 Oct. MedComm (2020). 2023. PMID: 37719444 Free PMC article. Review.
-
The Pyroptotic and Nonpyroptotic Roles of Gasdermins in Modulating Cancer Progression and Their Perspectives on Cancer Therapeutics.Arch Immunol Ther Exp (Warsz). 2023 May 31;71(1):14. doi: 10.1007/s00005-023-00678-9. Arch Immunol Ther Exp (Warsz). 2023. PMID: 37258998 Review.
-
Sec62 promotes gastric cancer metastasis through mediating UPR-induced autophagy activation.Cell Mol Life Sci. 2022 Feb 15;79(2):133. doi: 10.1007/s00018-022-04143-2. Cell Mol Life Sci. 2022. PMID: 35165763 Free PMC article.
-
Deep (phospho)proteomics profiling of pre- treatment needle biopsies identifies signatures of treatment resistance in HER2+ breast cancer.Cell Rep Med. 2023 Oct 17;4(10):101203. doi: 10.1016/j.xcrm.2023.101203. Epub 2023 Oct 3. Cell Rep Med. 2023. PMID: 37794585 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
