

Hepatitis B Virus Precore Protein p22 Inhibits Alpha Interferon Signaling by Blocking STAT Nuclear Translocation
- PMID: 31019054
- PMCID: PMC6580977
- DOI: 10.1128/JVI.00196-19
Hepatitis B Virus Precore Protein p22 Inhibits Alpha Interferon Signaling by Blocking STAT Nuclear Translocation
Abstract
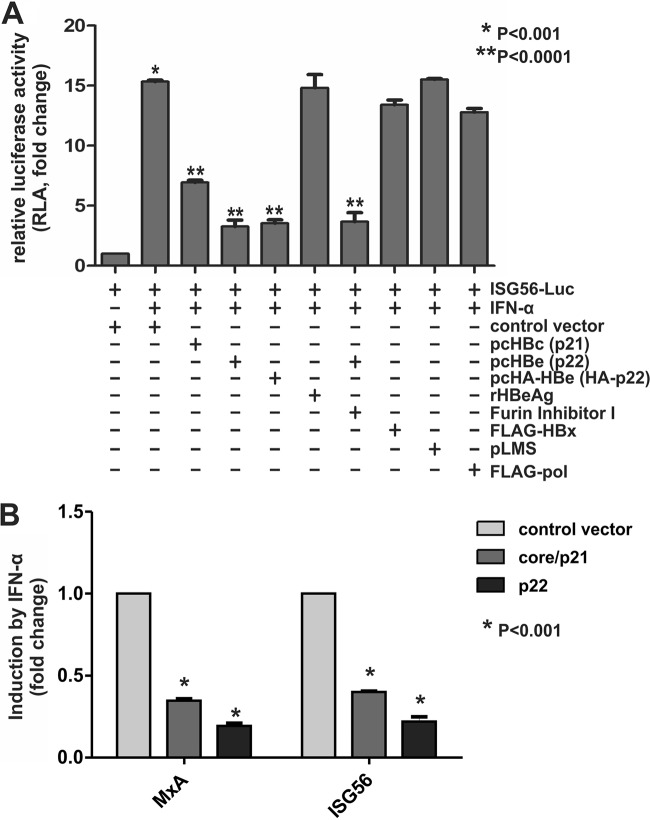
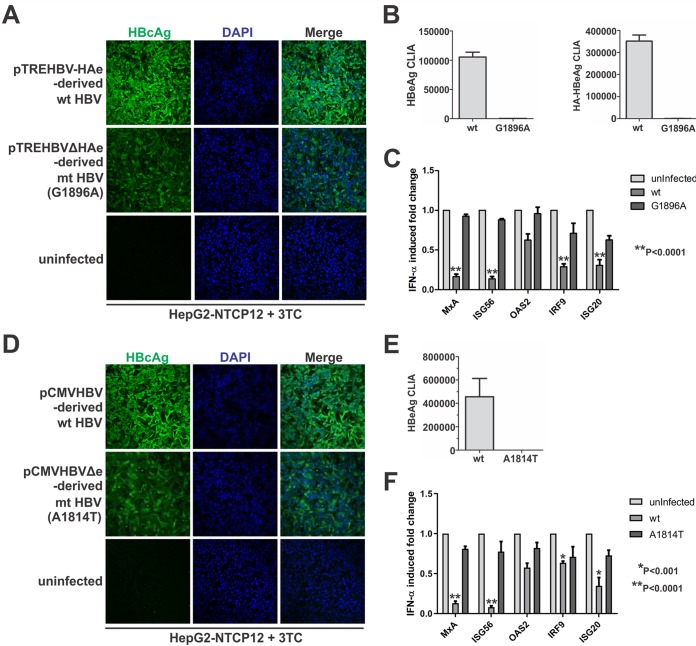
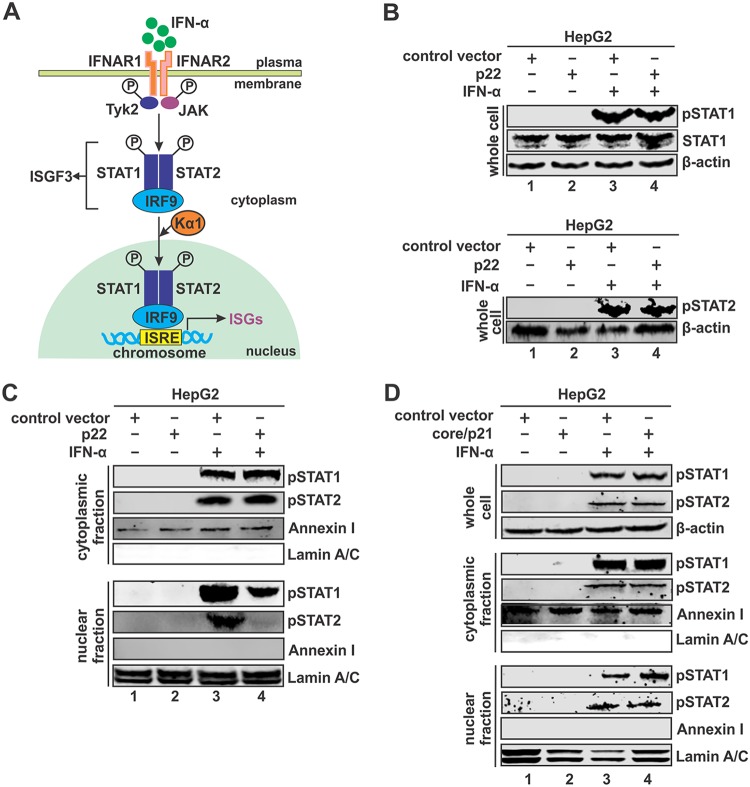
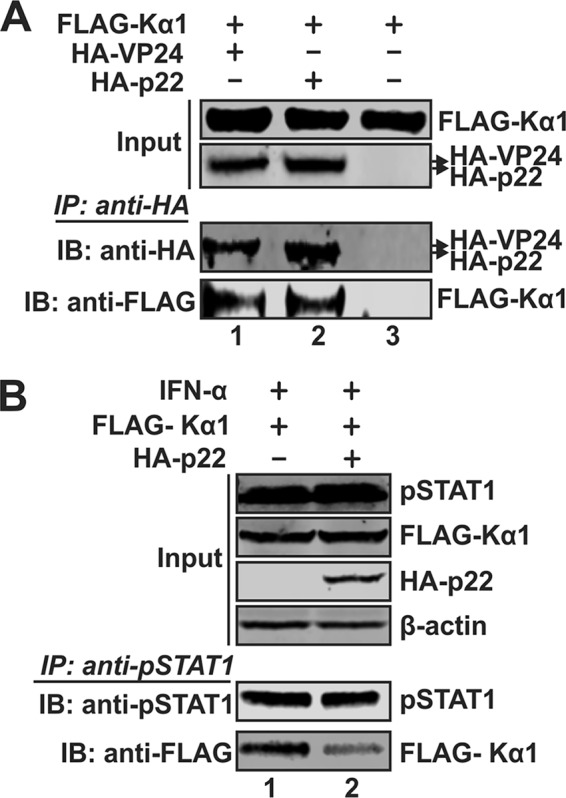
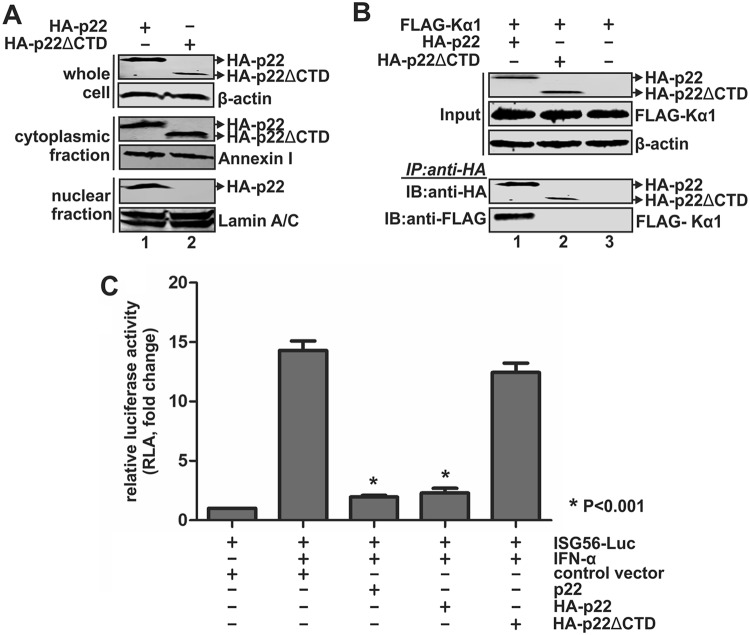
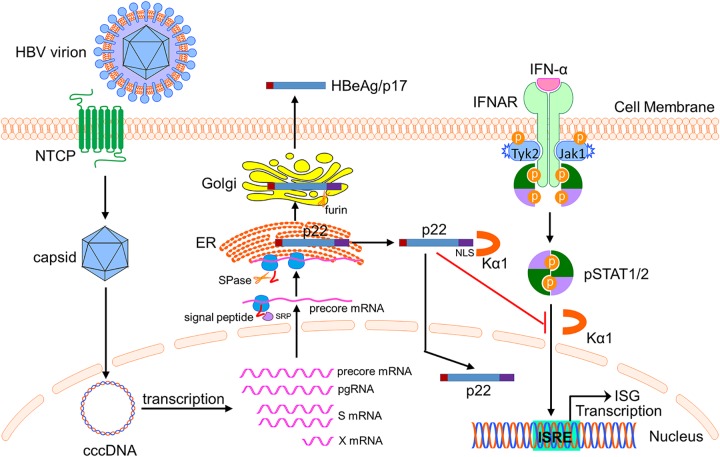
Antagonism of host immune defenses against hepatitis B virus (HBV) infection by the viral proteins is speculated to cause HBV persistence and the development of chronic hepatitis. The circulating hepatitis B e antigen (HBeAg, p17) is known to manipulate host immune responses to assist in the establishment of persistent viral infection, and HBeAg-positive (HBeAg+) patients respond less effectively to IFN-α therapy than do HBeAg-negative (HBeAg-) patients in clinical practice. However, the function(s) of the intracellular form of HBeAg, previously reported as the precore protein intermediate (p22) without the N-terminal signal peptide, remains elusive. Here, we report that the cytosolic p22 protein, but not the secreted HBeAg, significantly reduces interferon-stimulated response element (ISRE) activity and the expression of interferon-stimulated genes (ISGs) upon alpha interferon (IFN-α) stimulation in cell cultures. In line with this, HBeAg+ patients exhibit weaker induction of ISGs in their livers than do HBeAg- patients upon IFN-α therapy. Mechanistically, while p22 does not alter the total STAT1 or pSTAT1 levels in cells treated with IFN-α, it blocks the nuclear translocation of pSTAT1 by interacting with the nuclear transport factor karyopherin α1 through its C-terminal arginine-rich domain. In summary, our study suggests that HBV precore protein, specifically the p22 form, impedes JAK-STAT signaling to help the virus evade the host innate immune response and, thus, causes resistance to IFN therapy.IMPORTANCE Chronic hepatitis B virus (HBV) infection continues to be a major global health concern, and patients who fail to mount an efficient immune response to clear the virus will develop a life-long chronic infection that can progress to chronic active hepatitis, cirrhosis, and primary hepatocellular carcinoma. There is no definite cure for chronic hepatitis B, and alpha interferon (IFN-α) is the only available immunomodulatory drug, to which only a minority of chronic patients are responsive, with hepatitis B e antigen (HBeAg)-negative patients responding better than HBeAg-positive patients. We herein report that the intracellular HBeAg, also known as precore or p22, inhibits the antiviral signaling of IFN-α, which sheds light on the enigmatic function of precore protein in shaping HBV chronicity and provides a perspective toward areas that need to be further studied to make the current therapy better until a cure is achieved.
Keywords: hepatitis B virus; interferons.
Copyright © 2019 American Society for Microbiology.
Figures
Similar articles
-
Hepatitis B Virus e Antigen Activates the Suppressor of Cytokine Signaling 2 to Repress Interferon Action.Sci Rep. 2017 May 11;7(1):1729. doi: 10.1038/s41598-017-01773-6. Sci Rep. 2017. PMID: 28496097 Free PMC article.
-
In vitro expression of precore proteins of hepatitis B virus subgenotype A1 is affected by HBcAg, and can affect HBsAg secretion.Sci Rep. 2021 Apr 14;11(1):8167. doi: 10.1038/s41598-021-87529-9. Sci Rep. 2021. PMID: 33854155 Free PMC article.
-
Downregulation of interleukin-18-mediated cell signaling and interferon gamma expression by the hepatitis B virus e antigen.J Virol. 2014 Sep;88(18):10412-20. doi: 10.1128/JVI.00111-14. Epub 2014 May 28. J Virol. 2014. PMID: 24872585 Free PMC article.
-
Treatment of chronic hepatitis B: Evolution over two decades.J Gastroenterol Hepatol. 2011 Jan;26 Suppl 1:138-43. doi: 10.1111/j.1440-1746.2010.06545.x. J Gastroenterol Hepatol. 2011. PMID: 21199525 Review.
-
The Complex Role of HBeAg and Its Precursors in the Pathway to Hepatocellular Carcinoma.Viruses. 2023 Mar 27;15(4):857. doi: 10.3390/v15040857. Viruses. 2023. PMID: 37112837 Free PMC article. Review.
Cited by
-
Tripartite Motif-Containing Protein 65 (TRIM65) Inhibits Hepatitis B Virus Transcription.Viruses. 2024 May 31;16(6):890. doi: 10.3390/v16060890. Viruses. 2024. PMID: 38932182 Free PMC article.
-
MafF Is an Antiviral Host Factor That Suppresses Transcription from Hepatitis B Virus Core Promoter.J Virol. 2021 Jul 12;95(15):e0076721. doi: 10.1128/JVI.00767-21. Epub 2021 Jul 12. J Virol. 2021. PMID: 33980595 Free PMC article.
-
Identification of a Novel HBV Encoded miRNA Using Next Generation Sequencing.Viruses. 2022 Jun 5;14(6):1223. doi: 10.3390/v14061223. Viruses. 2022. PMID: 35746694 Free PMC article.
-
Proteomic Analysis of Nuclear Hepatitis B Virus Relaxed Circular DNA-Associated Proteins Identifies UV-Damaged DNA Binding Protein as a Host Factor Involved in Covalently Closed Circular DNA Formation.J Virol. 2022 Jan 26;96(2):e0136021. doi: 10.1128/JVI.01360-21. Epub 2021 Oct 27. J Virol. 2022. PMID: 34705558 Free PMC article.
-
Interferon and Hepatitis B: Current and Future Perspectives.Front Immunol. 2021 Sep 7;12:733364. doi: 10.3389/fimmu.2021.733364. eCollection 2021. Front Immunol. 2021. PMID: 34557195 Free PMC article. Review.
References
-
- Alter H, Block T, Brown N, Brownstein A, Brosgart C, Chang KM, Chen PJ, Chisari FV, Cohen C, El-Serag H, Feld J, Gish R, Glenn J, Greten T, Guo H, Guo JT, Hoshida Y, Hu J, Kowdley KV, Li W, Liang J, Locarnini S, Lok AS, Mason W, McMahon B, Mehta A, Perrillo R, Revill P, Rice CM, Rinaudo J, Schinazi R, Seeger C, Shetty K, Tavis J, Zoulim F. 2018. A research agenda for curing chronic hepatitis B virus infection. Hepatology 67:1127–1131. doi:10.1002/hep.29509. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
