

Site-specific glycation of Aβ1-42 affects fibril formation and is neurotoxic
- PMID: 30996005
- PMCID: PMC6552435
- DOI: 10.1074/jbc.RA118.006846
Site-specific glycation of Aβ1-42 affects fibril formation and is neurotoxic
Abstract
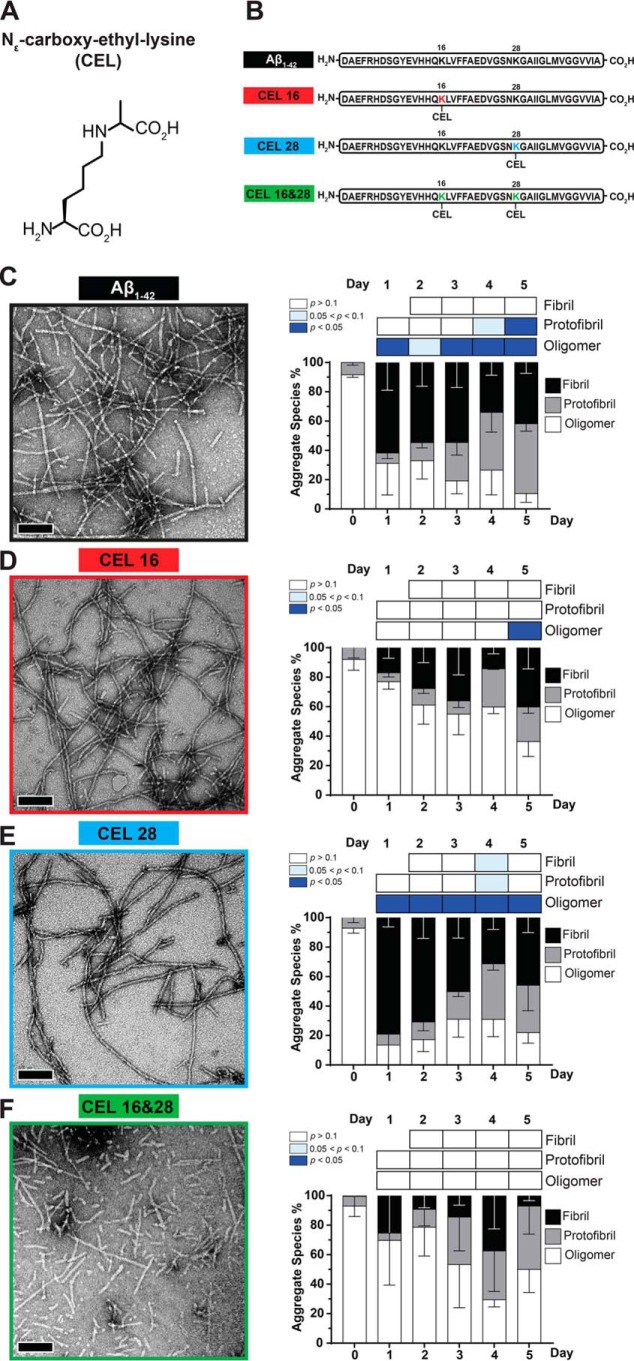
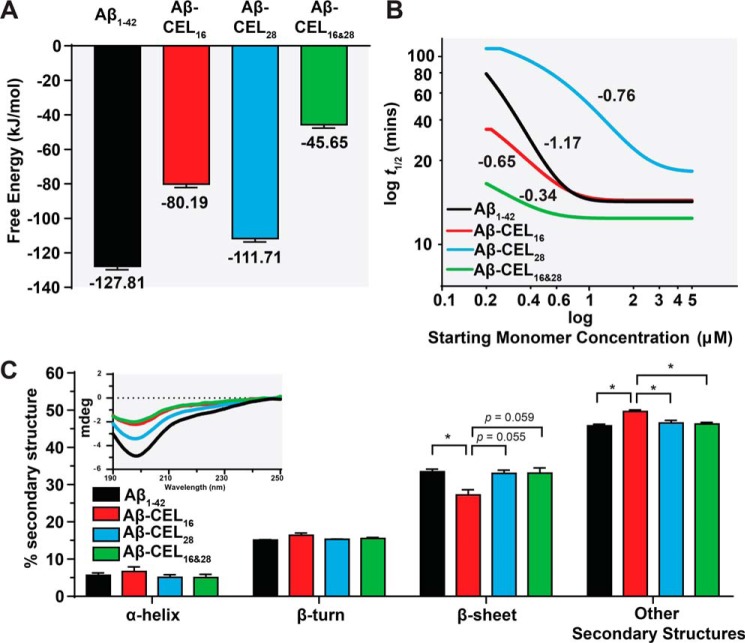
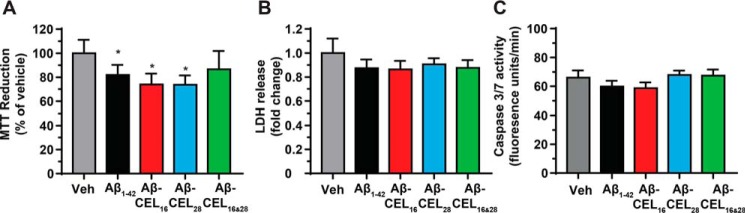
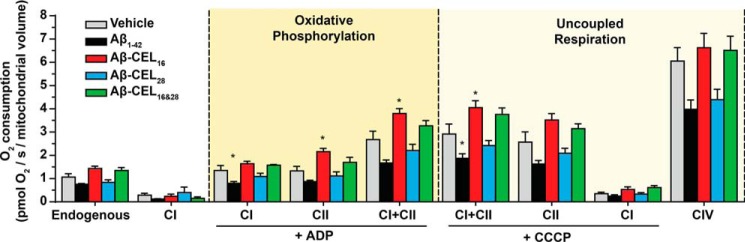
Aβ1-42 is involved in Alzheimer's disease (AD) pathogenesis and is prone to glycation, an irreversible process where proteins accumulate advanced glycated end products (AGEs). Nϵ-(Carboxyethyl)lysine (CEL) is a common AGE associated with AD patients and occurs at either Lys-16 or Lys-28 of Aβ1-42. Methyglyoxal is commonly used for the unspecific glycation of Aβ1-42, which results in a complex mixture of AGE-modified peptides and makes interpretation of a causative AGE at a specific amino acid residue difficult. We address this issue by chemically synthesizing defined CEL modifications on Aβ1-42 at Lys-16 (Aβ-CEL16), Lys-28 (Aβ-CEL28), and Lys-16 and -28 (Aβ-CEL16&28). We demonstrated that double-CEL glycations at Lys-16 and Lys-28 of Aβ1-42 had the most profound impact on the ability to form amyloid fibrils. In silico predictions indicated that Aβ-CEL16&28 had a substantial decrease in free energy change, which contributes to fibril destabilization, and a increased aggregation rate. Single-CEL glycations at Lys-28 of Aβ1-42 had the least impact on fibril formation, whereas CEL glycations at Lys-16 of Aβ1-42 delayed fibril formation. We also tested these peptides for neuronal toxicity and mitochondrial function on a retinoic acid-differentiated SH-SY5Y human neuroblastoma cell line (RA-differentiated SH-SY5Y). Only Aβ-CEL16 and Aβ-CEL28 were neurotoxic, possibly through a nonmitochondrial pathway, whereas Aβ-CEL16&28 showed no neurotoxicity. Interestingly, Aβ-CEL16&28 had depolarized the mitochondrial membrane potential, whereas Aβ-CEL16 had increased mitochondrial respiration at complex II. These results may indicate mitophagy or an alternate route of metabolism, respectively. Therefore, our results provides insight into potential therapeutic approaches against neurotoxic CEL-glycated Aβ1-42.
Keywords: Alzheimer disease; N-ϵ-(carboxyethyl)lysine (CEL); advanced glycated end product (AGE); amyloid-β (Aβ); fibril; glycation; mitochondria; neurodegeneration; protein aggregation.
© 2019 Ng et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures

Similar articles
-
Amyloid beta 1-40 and 1-42 fibril ratios and maturation level cause conformational differences with minimal impact on autophagy and cytotoxicity.J Neurochem. 2024 Sep;168(9):3308-3322. doi: 10.1111/jnc.16201. Epub 2024 Aug 12. J Neurochem. 2024. PMID: 39133499
-
Binding of Cu2+ to Aβ1-29 causes aggregation and toxicity in SH-SY5Y cells.Biochem Biophys Res Commun. 2021 Jan 1;534:617-623. doi: 10.1016/j.bbrc.2020.11.031. Epub 2020 Nov 15. Biochem Biophys Res Commun. 2021. PMID: 33208229
-
Autophagy Activation Alleviates Amyloid-β-Induced Oxidative Stress, Apoptosis and Neurotoxicity in Human Neuroblastoma SH-SY5Y Cells.Neurotox Res. 2017 Oct;32(3):351-361. doi: 10.1007/s12640-017-9746-5. Epub 2017 May 8. Neurotox Res. 2017. PMID: 28484969
-
Medicinal Chemistry Focusing on Aggregation of Amyloid-β.Chem Pharm Bull (Tokyo). 2016;64(1):1-7. doi: 10.1248/cpb.c15-00742. Chem Pharm Bull (Tokyo). 2016. PMID: 26726739 Review.
-
[Cell-surface Initial Aggregation Process of Amyloid-β Peptides Studied by Fluorescence Correlation Spectroscopy].Yakugaku Zasshi. 2021;141(6):825-829. doi: 10.1248/yakushi.20-00251-2. Yakugaku Zasshi. 2021. PMID: 34078789 Review. Japanese.
Cited by
-
Reduced Influence of apoE on Aβ43 Aggregation and Reduced Vascular Aβ43 Toxicity as Compared with Aβ40 and Aβ42.Mol Neurobiol. 2020 Apr;57(4):2131-2141. doi: 10.1007/s12035-020-01873-x. Epub 2020 Jan 17. Mol Neurobiol. 2020. PMID: 31953617 Free PMC article.
-
Inhibition of neuroinflammatory nitric oxide signaling suppresses glycation and prevents neuronal dysfunction in mouse prion disease.Proc Natl Acad Sci U S A. 2021 Mar 9;118(10):e2009579118. doi: 10.1073/pnas.2009579118. Proc Natl Acad Sci U S A. 2021. PMID: 33653950 Free PMC article.
-
An overview on glycation: molecular mechanisms, impact on proteins, pathogenesis, and inhibition.Biophys Rev. 2024 Apr 12;16(2):189-218. doi: 10.1007/s12551-024-01188-4. eCollection 2024 Apr. Biophys Rev. 2024. PMID: 38737201 Free PMC article. Review.
-
Stability of Protein Pharmaceuticals: Recent Advances.Pharm Res. 2024 Jul;41(7):1301-1367. doi: 10.1007/s11095-024-03726-x. Epub 2024 Jun 27. Pharm Res. 2024. PMID: 38937372 Review.
-
SIRT7 Deficiency Protects against Aβ42-Induced Apoptosis through the Regulation of NOX4-Derived Reactive Oxygen Species Production in SH-SY5Y Cells.Int J Mol Sci. 2022 Aug 12;23(16):9027. doi: 10.3390/ijms23169027. Int J Mol Sci. 2022. PMID: 36012298 Free PMC article.
References
-
- Sasaki N., Fukatsu R., Tsuzuki K., Hayashi Y., Yoshida T., Fujii N., Koike T., Wakayama I., Yanagihara R., Garruto R., Amano N., and Makita Z. (1998) Advanced glycation end products in Alzheimer's disease and other neurodegenerative diseases. Am. J. Pathol. 153, 1149–1155 10.1016/S0002-9440(10)65659-3 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
