

Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes
- PMID: 30987118
- PMCID: PMC6480154
- DOI: 10.3390/ijms20071658
Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes
Abstract
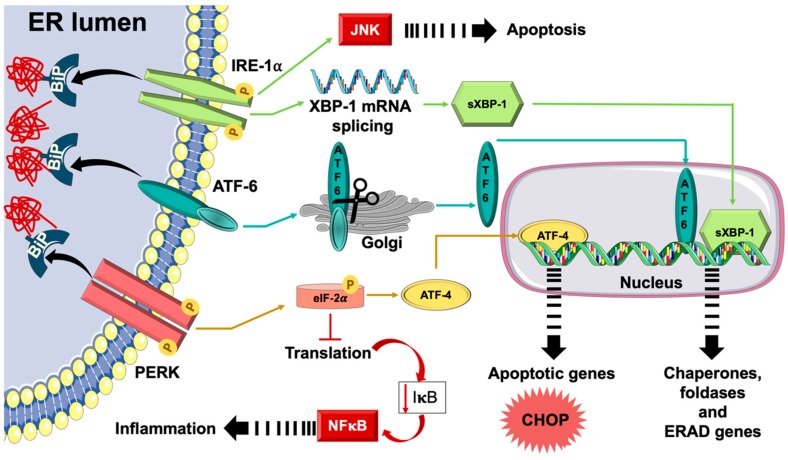
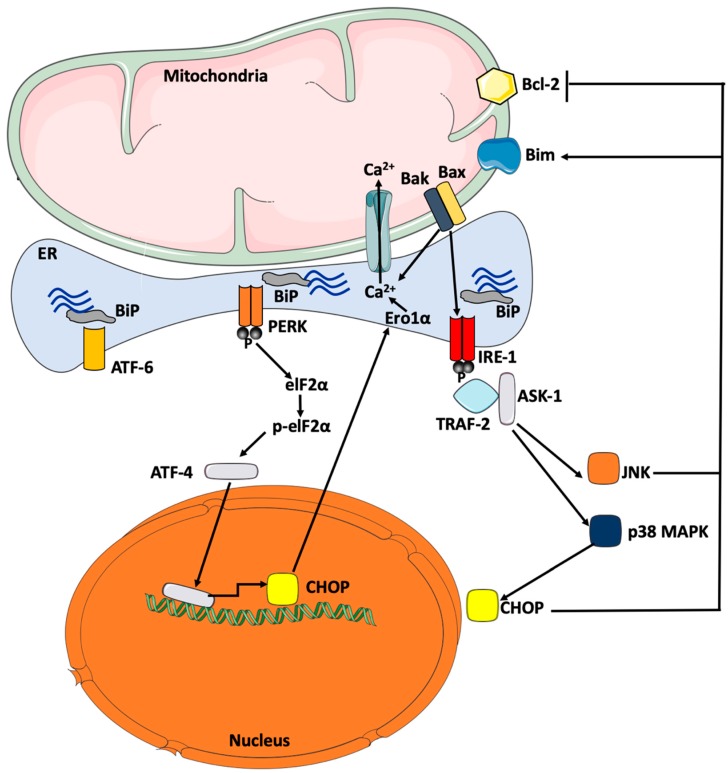
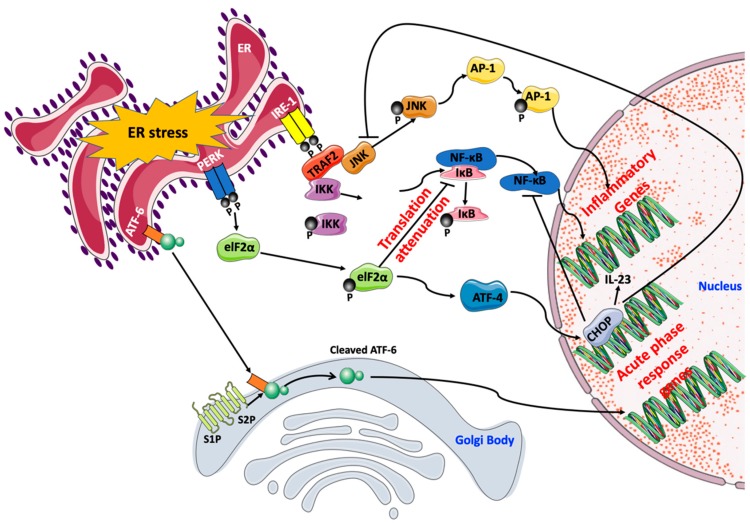
Physical inactivity and sedentary lifestyle contribute to the widespread epidemic of obesity among both adults and children leading to rising cases of diabetes. Cardiovascular disease complications associated with obesity and diabetes are closely linked to insulin resistance and its complex implications on vascular cells particularly endothelial cells. Endoplasmic reticulum (ER) stress is activated following disruption in post-translational protein folding and maturation within the ER in metabolic conditions characterized by heavy demand on protein synthesis, such as obesity and diabetes. ER stress has gained much interest as a key bridging and converging molecular link between insulin resistance, oxidative stress, and endothelial cell dysfunction and, hence, represents an interesting drug target for diabetes and its cardiovascular complications. We reviewed here the role of ER stress in endothelial cell dysfunction, the primary step in the onset of atherosclerosis and cardiovascular disease. We specifically focused on the contribution of oxidative stress, insulin resistance, endothelial cell death, and cellular inflammation caused by ER stress in endothelial cell dysfunction and the process of atherogenesis.
Keywords: atherosclerosis; diabetes; endoplasmic reticulum stress; endothelial dysfunction; unfolded protein response.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Endoplasmic reticulum stress and the development of endothelial dysfunction.Am J Physiol Heart Circ Physiol. 2017 Mar 1;312(3):H355-H367. doi: 10.1152/ajpheart.00437.2016. Epub 2016 Dec 6. Am J Physiol Heart Circ Physiol. 2017. PMID: 27923788 Review.
-
Reversibility of endothelial dysfunction in diabetes: role of polyphenols.Br J Nutr. 2016 Jul;116(2):223-46. doi: 10.1017/S0007114516001884. Epub 2016 Jun 6. Br J Nutr. 2016. PMID: 27264638 Review.
-
Liraglutide Treatment Reduces Endothelial Endoplasmic Reticulum Stress and Insulin Resistance in Patients With Diabetes Mellitus.J Am Heart Assoc. 2018 Sep 18;7(18):e009379. doi: 10.1161/JAHA.118.009379. J Am Heart Assoc. 2018. PMID: 30371206 Free PMC article.
-
Role of endoplasmic reticulum stress in endothelial dysfunction.Nutr Metab Cardiovasc Dis. 2016 Oct;26(10):863-71. doi: 10.1016/j.numecd.2016.05.008. Epub 2016 May 28. Nutr Metab Cardiovasc Dis. 2016. PMID: 27345757 Review.
-
Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis.Diab Vasc Dis Res. 2017 Jan;14(1):14-23. doi: 10.1177/1479164116666762. Epub 2016 Oct 20. Diab Vasc Dis Res. 2017. PMID: 27941052 Free PMC article. Review.
Cited by
-
Endothelial Unfolded Protein Response-Mediated Cytoskeletal Effects.Cell Biochem Funct. 2024 Dec;42(8):e70007. doi: 10.1002/cbf.70007. Cell Biochem Funct. 2024. PMID: 39449673 Review.
-
Liquid-Liquid Phase Separation in Cardiovascular Diseases.Cells. 2022 Sep 28;11(19):3040. doi: 10.3390/cells11193040. Cells. 2022. PMID: 36231002 Free PMC article. Review.
-
Calciprotein Particles Induce Cellular Compartment-Specific Proteome Alterations in Human Arterial Endothelial Cells.J Cardiovasc Dev Dis. 2023 Dec 22;11(1):5. doi: 10.3390/jcdd11010005. J Cardiovasc Dev Dis. 2023. PMID: 38248875 Free PMC article.
-
Hemophagocytic lymphohistiocytosis as an onset of diffuse large B-cell lymphoma: A case report.Oncol Lett. 2022 Jul 5;24(3):298. doi: 10.3892/ol.2022.13418. eCollection 2022 Sep. Oncol Lett. 2022. PMID: 35949601 Free PMC article.
-
Current Status of Endoplasmic Reticulum Stress in Type II Diabetes.Molecules. 2021 Jul 19;26(14):4362. doi: 10.3390/molecules26144362. Molecules. 2021. PMID: 34299638 Free PMC article. Review.
References
-
- McGill H.C., Jr., McMahan C.A., Herderick E.E., Zieske A.W., Malcom G.T., Tracy R.E., Strong J.P., Pathobiological Determinants of Atherosclerosis in Youth Research Group Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation. 2002;105:2712–2718. doi: 10.1161/01.CIR.0000018121.67607.CE. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
