

p53-TP53-Induced Glycolysis Regulator Mediated Glycolytic Suppression Attenuates DNA Damage and Genomic Instability in Fanconi Anemia Hematopoietic Stem Cells
- PMID: 30977208
- PMCID: PMC6599562
- DOI: 10.1002/stem.3015
p53-TP53-Induced Glycolysis Regulator Mediated Glycolytic Suppression Attenuates DNA Damage and Genomic Instability in Fanconi Anemia Hematopoietic Stem Cells
Abstract
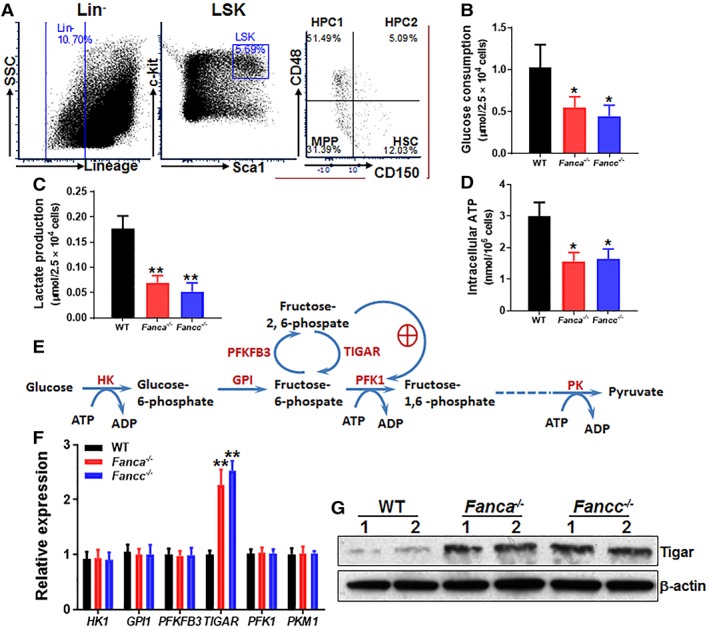
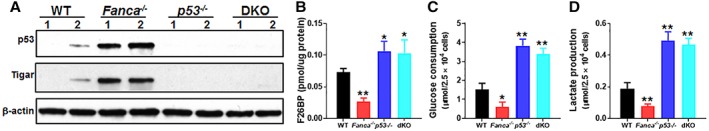
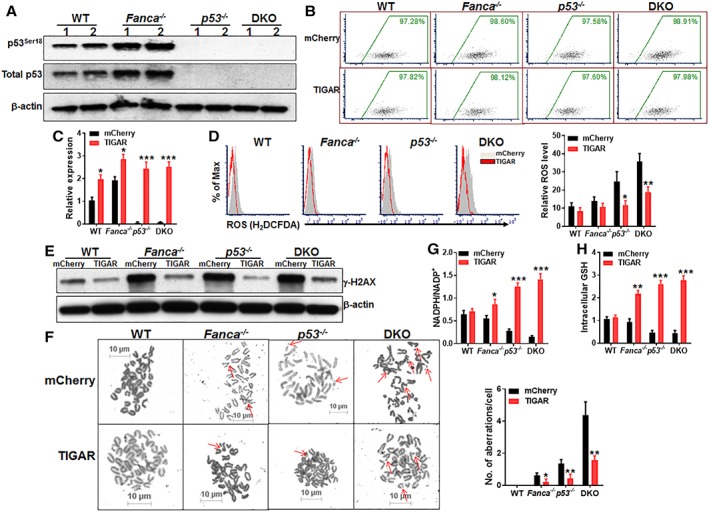
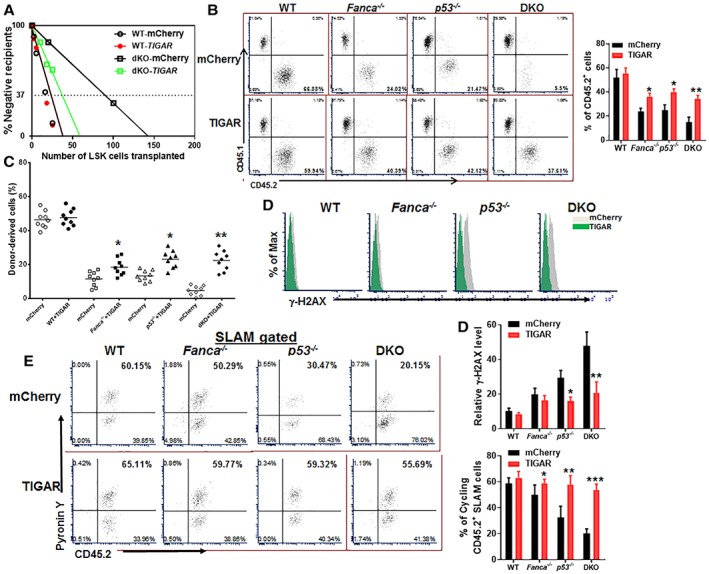
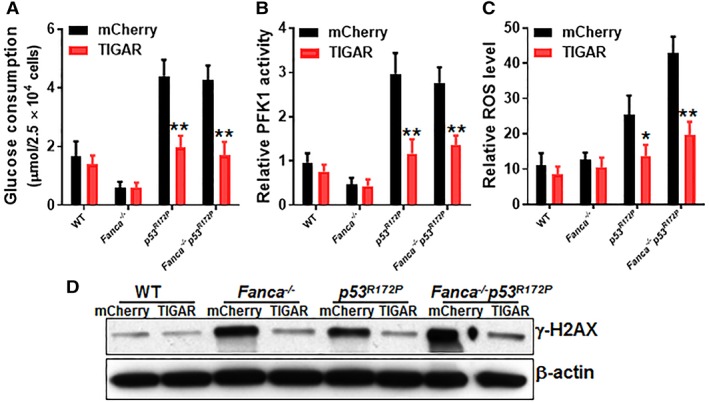
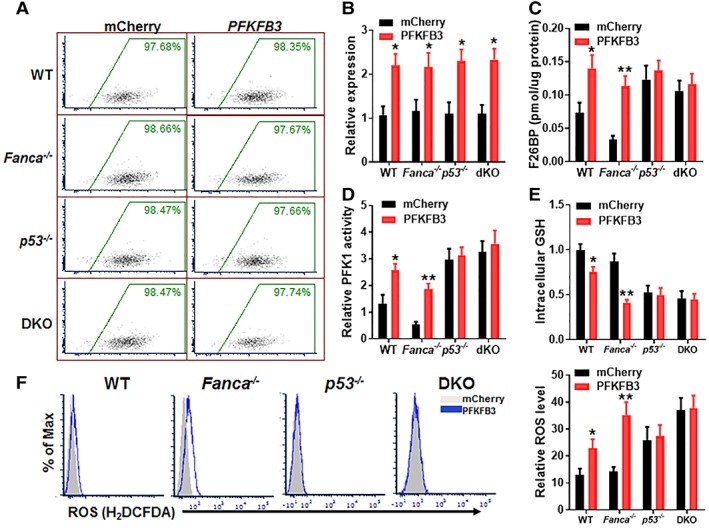
Emerging evidence has shown that resting quiescent hematopoietic stem cells (HSCs) prefer to utilize anaerobic glycolysis rather than mitochondrial respiration for energy production. Compelling evidence has also revealed that altered metabolic energetics in HSCs underlies the onset of certain blood diseases; however, the mechanisms responsible for energetic reprogramming remain elusive. We recently found that Fanconi anemia (FA) HSCs in their resting state are more dependent on mitochondrial respiration for energy metabolism than on glycolysis. In the present study, we investigated the role of deficient glycolysis in FA HSC maintenance. We observed significantly reduced glucose consumption, lactate production, and ATP production in HSCs but not in the less primitive multipotent progenitors or restricted hematopoietic progenitors of Fanca-/- and Fancc-/- mice compared with that of wild-type mice, which was associated with an overactivated p53 and TP53-induced glycolysis regulator, the TIGAR-mediated metabolic axis. We utilized Fanca-/- HSCs deficient for p53 to show that the p53-TIGAR axis suppressed glycolysis in FA HSCs, leading to enhanced pentose phosphate pathway and cellular antioxidant function and, consequently, reduced DNA damage and attenuated HSC exhaustion. Furthermore, by using Fanca-/- HSCs carrying the separation-of-function mutant p53R172P transgene that selectively impairs the p53 function in apoptosis but not cell-cycle control, we demonstrated that the cell-cycle function of p53 was not required for glycolytic suppression in FA HSCs. Finally, ectopic expression of the glycolytic rate-limiting enzyme PFKFB3 specifically antagonized p53-TIGAR-mediated metabolic reprogramming in FA HSCs. Together, our results suggest that p53-TIGAR metabolic axis-mediated glycolytic suppression may play a compensatory role in attenuating DNA damage and proliferative exhaustion in FA HSCs. Stem Cells 2019;37:937-947.
Keywords: Fanconi anemia; Glycolysis; Hematopoietic stem cells; TP53-inducible glycolysis and apoptosis regulator; p53.
© 2019 The Authors. Stem Cells published by Wiley Periodicals, Inc. on behalf of AlphaMed Press 2019.
Conflict of interest statement
The authors indicated no potential conflicts of interest.
Figures
Similar articles
-
SCO2 Mediates Oxidative Stress-Induced Glycolysis to Oxidative Phosphorylation Switch in Hematopoietic Stem Cells.Stem Cells. 2016 Apr;34(4):960-71. doi: 10.1002/stem.2260. Epub 2015 Dec 31. Stem Cells. 2016. PMID: 26676373 Free PMC article.
-
Cell-Cycle-Specific Function of p53 in Fanconi Anemia Hematopoietic Stem and Progenitor Cell Proliferation.Stem Cell Reports. 2018 Feb 13;10(2):339-346. doi: 10.1016/j.stemcr.2017.12.006. Epub 2018 Jan 4. Stem Cell Reports. 2018. PMID: 29307578 Free PMC article.
-
The non-homologous end-joining activity is required for Fanconi anemia fetal HSC maintenance.Stem Cell Res Ther. 2019 Mar 29;10(1):114. doi: 10.1186/s13287-019-1206-0. Stem Cell Res Ther. 2019. PMID: 30925933 Free PMC article.
-
Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor.Oncotarget. 2011 Dec;2(12):948-57. doi: 10.18632/oncotarget.389. Oncotarget. 2011. PMID: 22248668 Free PMC article. Review.
-
DNA damage and repair in the hematopoietic system.Acta Biochim Biophys Sin (Shanghai). 2022 Jan 25;54(6):847-857. doi: 10.3724/abbs.2022053. Acta Biochim Biophys Sin (Shanghai). 2022. PMID: 35593466 Free PMC article. Review.
Cited by
-
The role of PFKFB3 in maintaining colorectal cancer cell proliferation and stemness.Mol Biol Rep. 2022 Oct;49(10):9877-9891. doi: 10.1007/s11033-022-07513-y. Epub 2022 May 12. Mol Biol Rep. 2022. PMID: 35553342 Review.
-
Structure, regulation, and biological functions of TIGAR and its role in diseases.Acta Pharmacol Sin. 2021 Oct;42(10):1547-1555. doi: 10.1038/s41401-020-00588-y. Epub 2021 Jan 28. Acta Pharmacol Sin. 2021. PMID: 33510458 Free PMC article. Review.
-
The role of ubiquitination and deubiquitination in cancer metabolism.Mol Cancer. 2020 Oct 1;19(1):146. doi: 10.1186/s12943-020-01262-x. Mol Cancer. 2020. PMID: 33004065 Free PMC article. Review.
-
FANCD2 and HES1 suppress inflammation-induced PPARɣ to prevent haematopoietic stem cell exhaustion.Br J Haematol. 2021 Feb;192(3):652-663. doi: 10.1111/bjh.17230. Epub 2020 Nov 22. Br J Haematol. 2021. PMID: 33222180 Free PMC article.
-
Umbilical Cord Blood-Derived Cells Can Reconstruct Hematopoiesis in an Aplastic Anemia Animal Model.Stem Cells Int. 2024 Aug 12;2024:4095268. doi: 10.1155/2024/4095268. eCollection 2024. Stem Cells Int. 2024. PMID: 39161367 Free PMC article.
References
-
- Bagby GC Jr. Genetic basis of Fanconi anemia. Curr Opin Hematol 2003;10:68–76. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
