

ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism
- PMID: 30867319
- PMCID: PMC6878763
- DOI: 10.1126/scitranslmed.aaq1238
ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism
Abstract
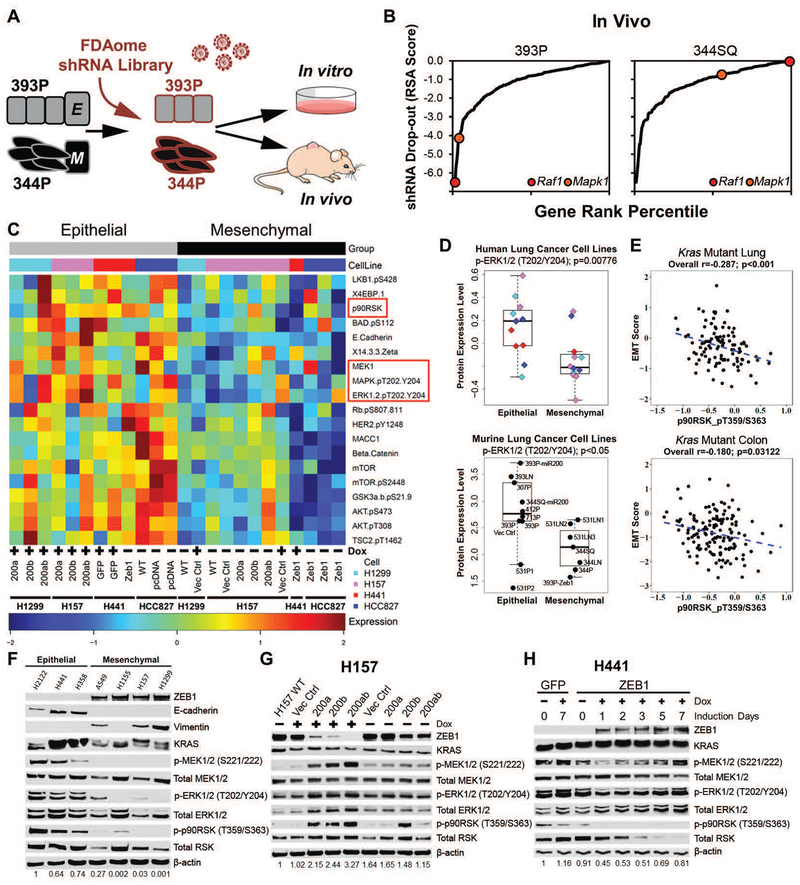
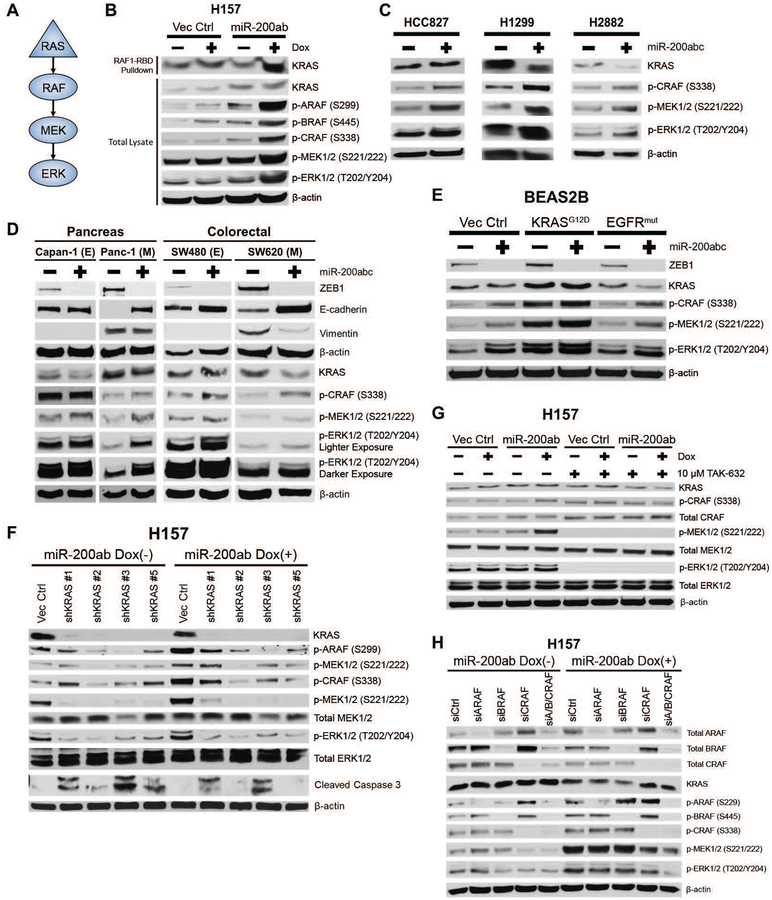
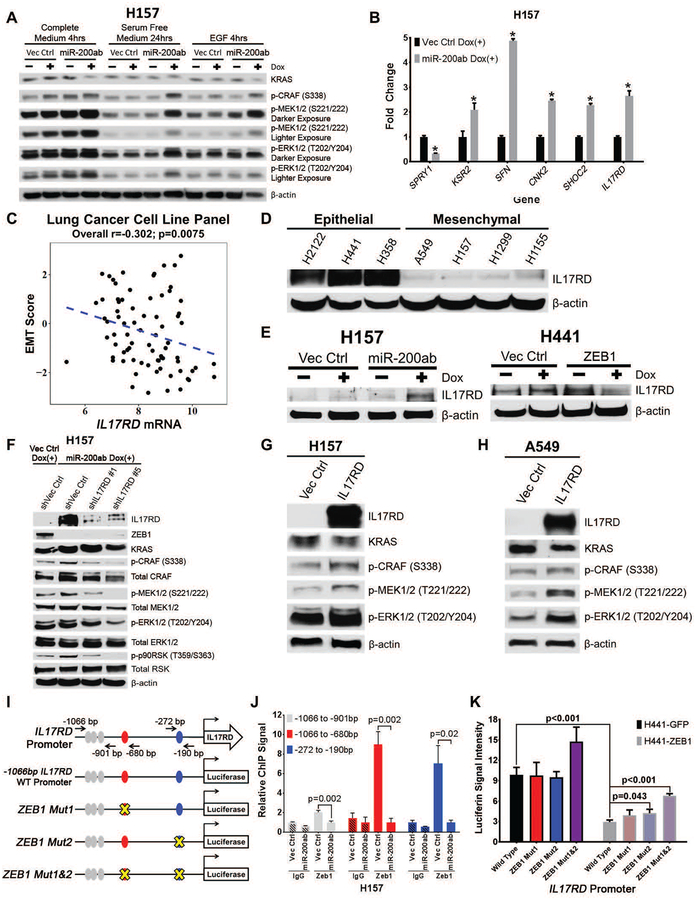
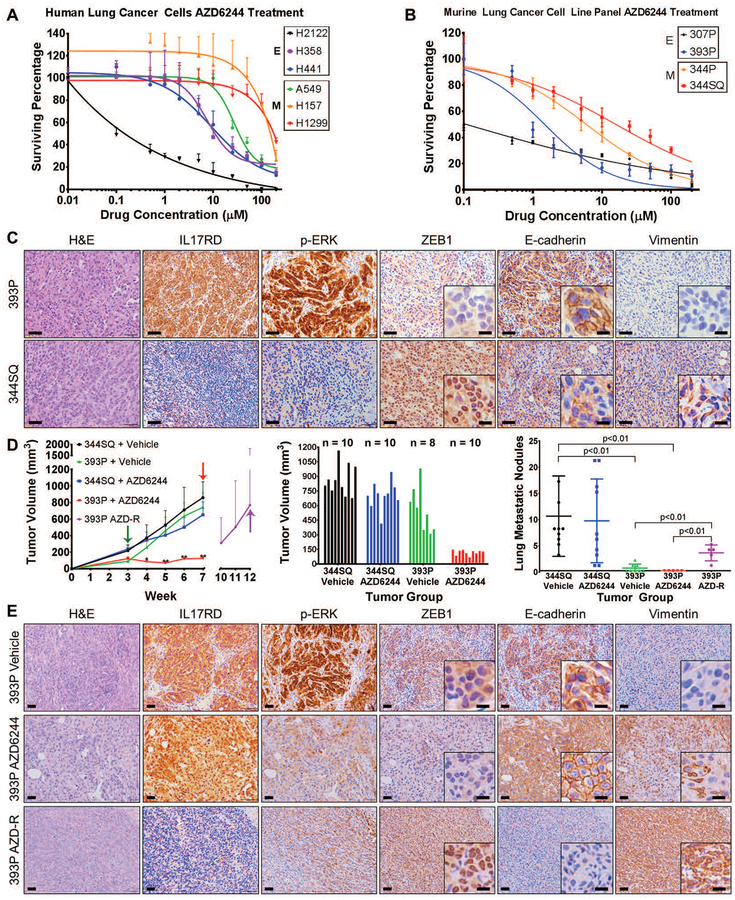
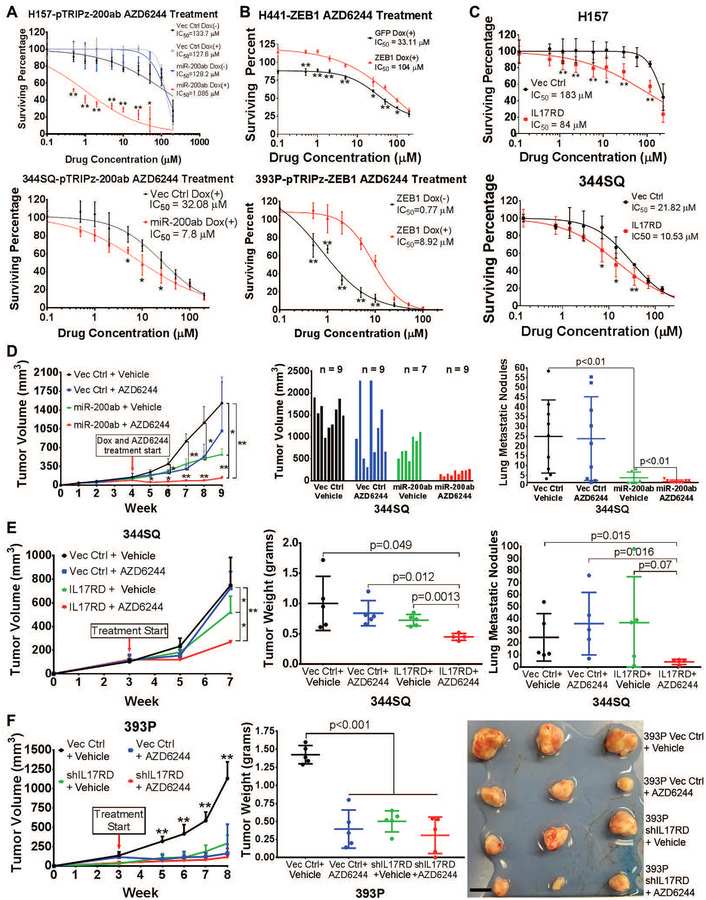
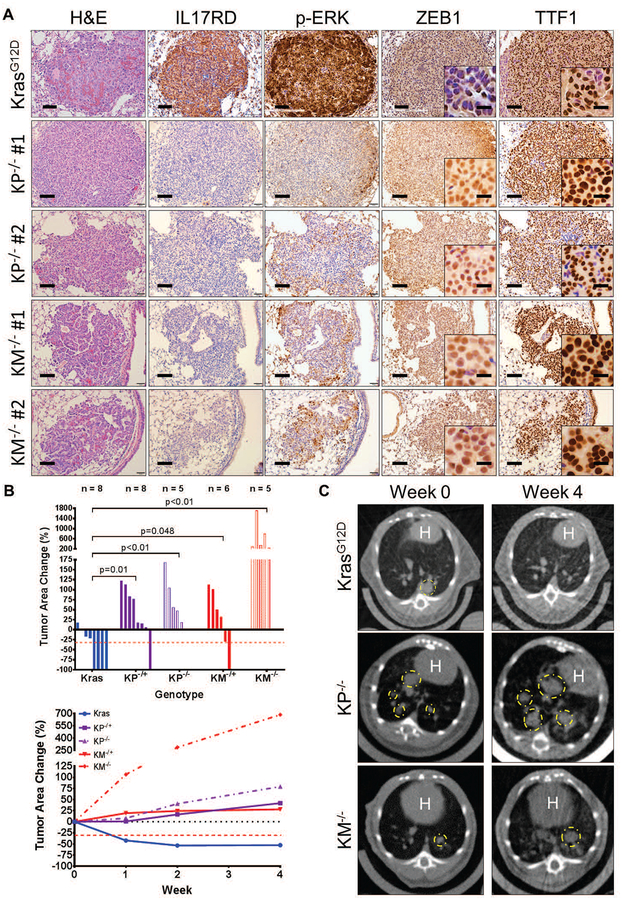
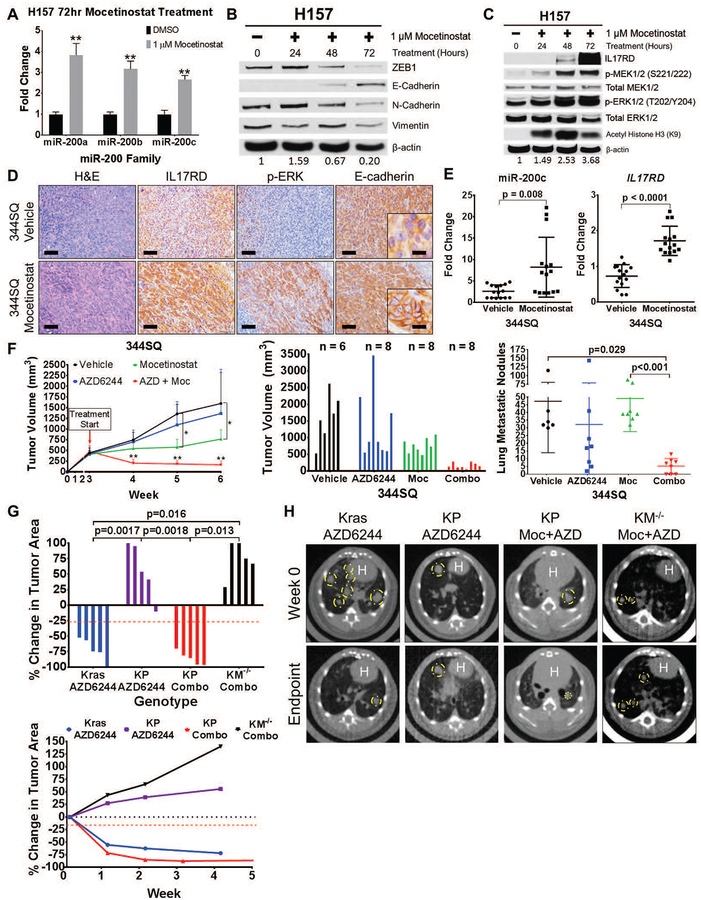
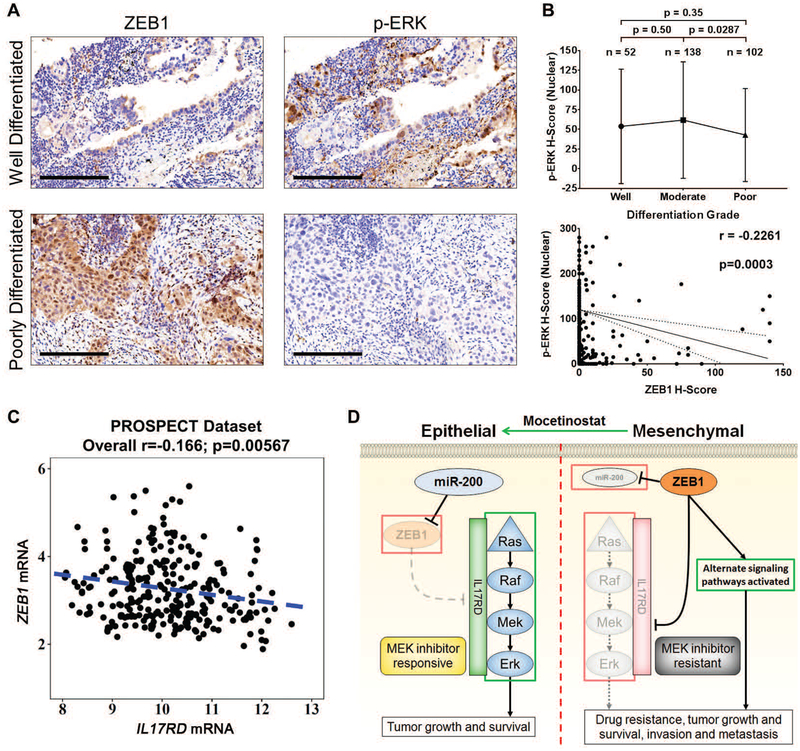
Mitogen-activated protein kinase (MAPK) kinase (MEK) inhibitors have failed to show clinical benefit in Kirsten rat sarcoma (KRAS) mutant lung cancer due to various resistance mechanisms. To identify differential therapeutic sensitivities between epithelial and mesenchymal lung tumors, we performed in vivo small hairpin RNA screens, proteomic profiling, and analysis of patient tumor datasets, which revealed an inverse correlation between mitogen-activated protein kinase (MAPK) signaling dependency and a zinc finger E-box binding homeobox 1 (ZEB1)-regulated epithelial-to-mesenchymal transition. Mechanistic studies determined that MAPK signaling dependency in epithelial lung cancer cells is due to the scaffold protein interleukin-17 receptor D (IL17RD), which is directly repressed by ZEB1. Lung tumors in multiple Kras mutant murine models with increased ZEB1 displayed low IL17RD expression, accompanied by MAPK-independent tumor growth and therapeutic resistance to MEK inhibition. Suppression of ZEB1 function with miR-200 expression or the histone deacetylase inhibitor mocetinostat sensitized resistant cancer cells to MEK inhibition and markedly reduced in vivo tumor growth, showing a promising combinatorial treatment strategy for KRAS mutant cancers. In human lung tumor samples, high ZEB1 and low IL17RD expression correlated with low MAPK signaling, presenting potential markers that predict patient response to MEK inhibitors.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Similar articles
-
An integrative pharmacogenomics analysis identifies therapeutic targets in KRAS-mutant lung cancer.EBioMedicine. 2019 Nov;49:106-117. doi: 10.1016/j.ebiom.2019.10.012. Epub 2019 Oct 23. EBioMedicine. 2019. PMID: 31668570 Free PMC article.
-
Th17 cells contribute to combination MEK inhibitor and anti-PD-L1 therapy resistance in KRAS/p53 mutant lung cancers.Nat Commun. 2021 May 10;12(1):2606. doi: 10.1038/s41467-021-22875-w. Nat Commun. 2021. PMID: 33972557 Free PMC article.
-
The protein kinase MAP3K19 phosphorylates MAP2Ks and thereby activates ERK and JNK kinases and increases viability of KRAS-mutant lung cancer cells.J Biol Chem. 2020 Jun 19;295(25):8470-8479. doi: 10.1074/jbc.RA119.012365. Epub 2020 Apr 30. J Biol Chem. 2020. PMID: 32358059 Free PMC article.
-
Key roles of EMT for adaptive resistance to MEK inhibitor in KRAS mutant lung cancer.Small GTPases. 2017 Jul 3;8(3):172-176. doi: 10.1080/21541248.2016.1210369. Epub 2016 Jul 8. Small GTPases. 2017. PMID: 27392325 Free PMC article. Review.
-
Resistance to MEK inhibitors: should we co-target upstream?Sci Signal. 2011 Mar 29;4(166):pe16. doi: 10.1126/scisignal.2001948. Sci Signal. 2011. PMID: 21447797 Review.
Cited by
-
KRAS mutation in pancreatic cancer.Semin Oncol. 2021 Feb;48(1):10-18. doi: 10.1053/j.seminoncol.2021.02.003. Epub 2021 Feb 23. Semin Oncol. 2021. PMID: 33676749 Free PMC article. Review.
-
Circ-HMGA2 (hsa_circ_0027446) promotes the metastasis and epithelial-mesenchymal transition of lung adenocarcinoma cells through the miR-1236-3p/ZEB1 axis.Cell Death Dis. 2021 Mar 24;12(4):313. doi: 10.1038/s41419-021-03601-2. Cell Death Dis. 2021. PMID: 33762580 Free PMC article.
-
Antitumor Activity of a Novel LAIR1 Antagonist in Combination with Anti-PD1 to Treat Collagen-Rich Solid Tumors.Mol Cancer Ther. 2024 Aug 1;23(8):1144-1158. doi: 10.1158/1535-7163.MCT-23-0866. Mol Cancer Ther. 2024. PMID: 38648067 Free PMC article.
-
Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients.Cell. 2020 Nov 12;183(4):850-859. doi: 10.1016/j.cell.2020.09.044. Epub 2020 Oct 15. Cell. 2020. PMID: 33065029 Free PMC article. Review.
-
Epithelial-mesenchymal transition: The history, regulatory mechanism, and cancer therapeutic opportunities.MedComm (2020). 2022 May 18;3(2):e144. doi: 10.1002/mco2.144. eCollection 2022 Jun. MedComm (2020). 2022. PMID: 35601657 Free PMC article. Review.
References
-
- Sharma SV, Haber DA, Settleman J, Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat Rev Cancer 10, 241–253 (2010). - PubMed
-
- Rodenhuis S, Slebos RJ, Clinical significance of ras oncogene activation in human lung cancer. Cancer Res 52, 2665s–2669s (1992). - PubMed
-
- Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, Haller A, Lothaire P, Meert AP, Noel S, Lafitte JJ, Sculier JP, The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. British journal of cancer 92, 131–139 (2005). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
