

Ablation of the calpain-targeted site in cardiac myosin binding protein-C is cardioprotective during ischemia-reperfusion injury
- PMID: 30862451
- PMCID: PMC7222036
- DOI: 10.1016/j.yjmcc.2019.03.006
Ablation of the calpain-targeted site in cardiac myosin binding protein-C is cardioprotective during ischemia-reperfusion injury
Abstract
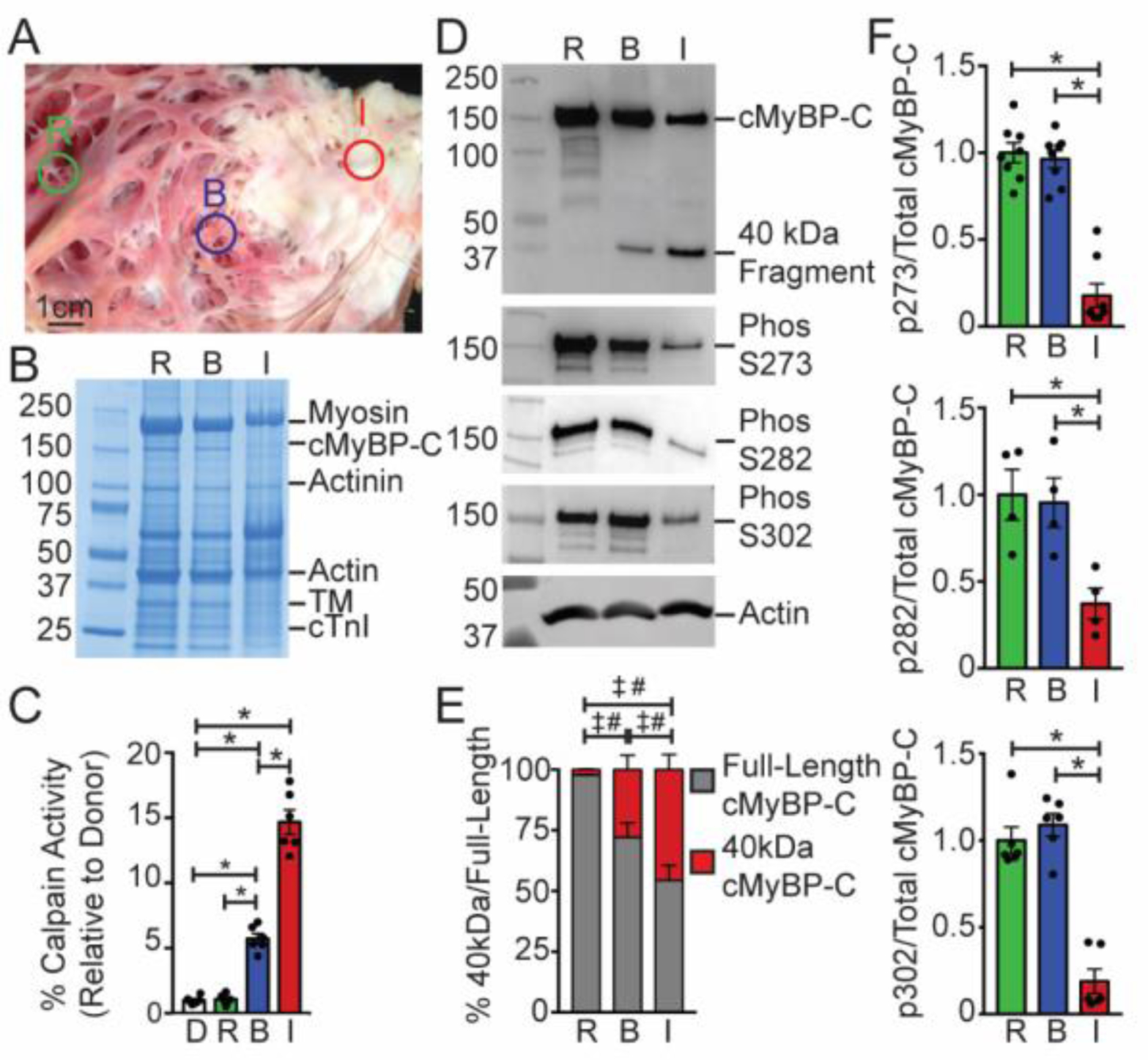
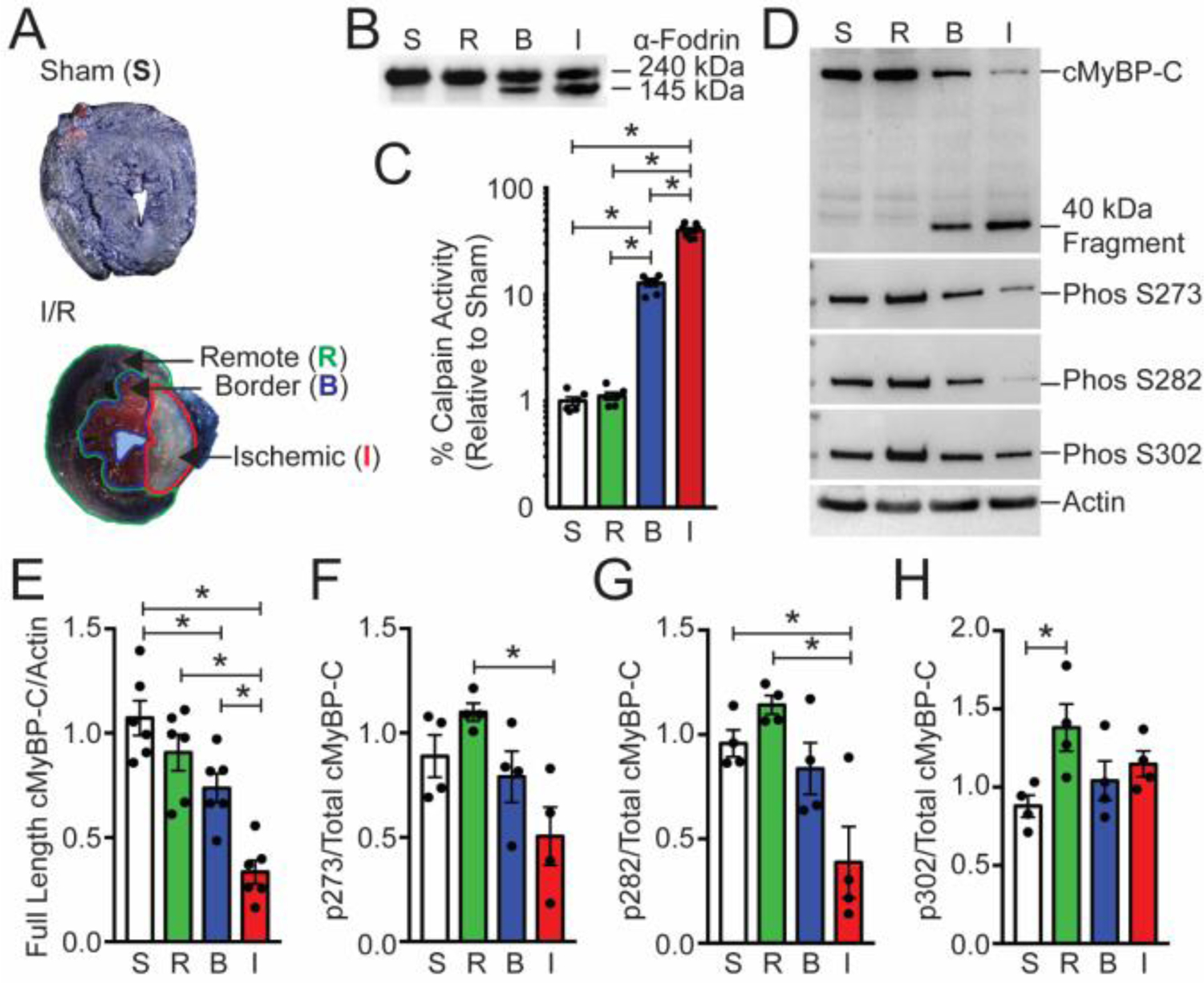
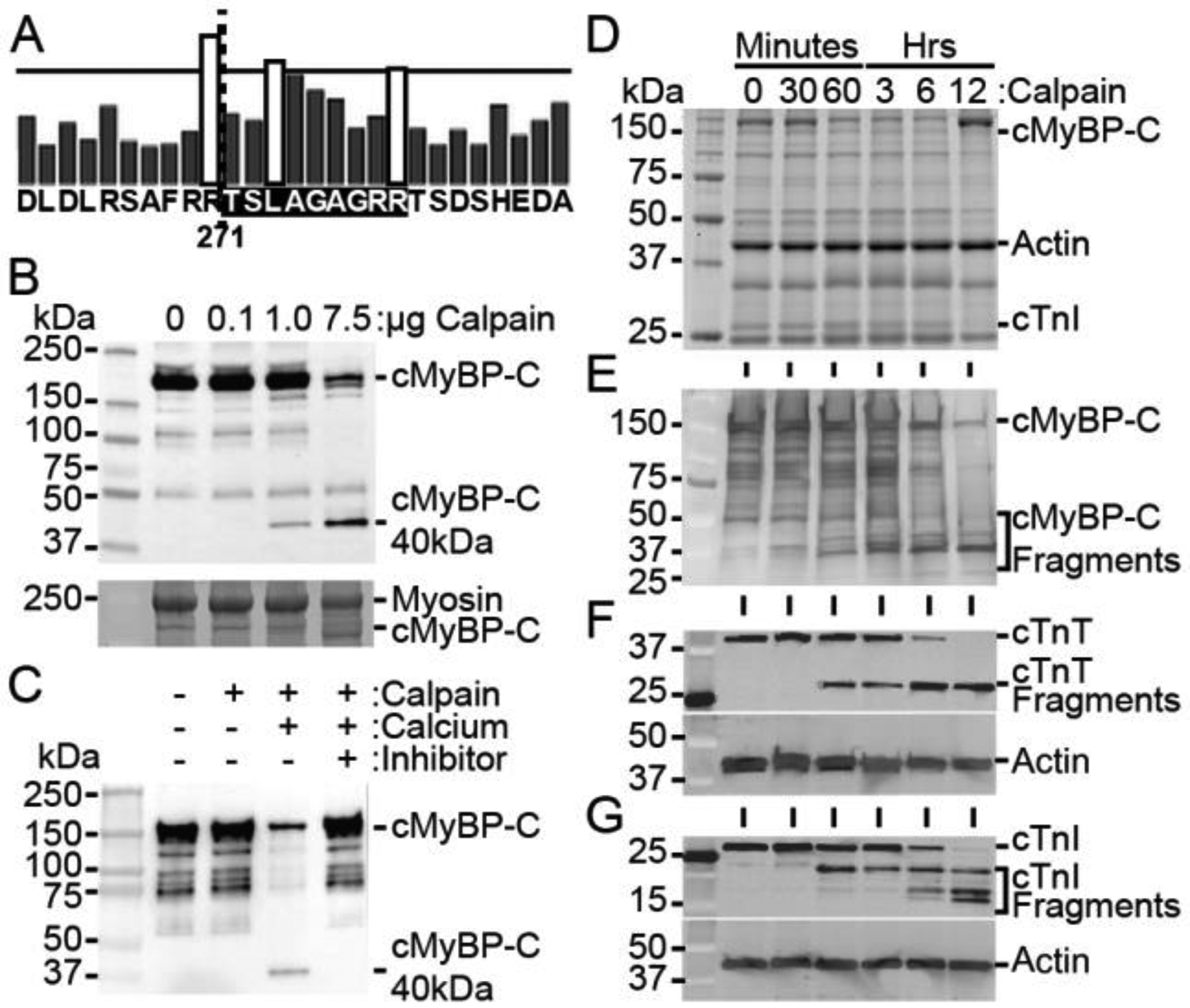
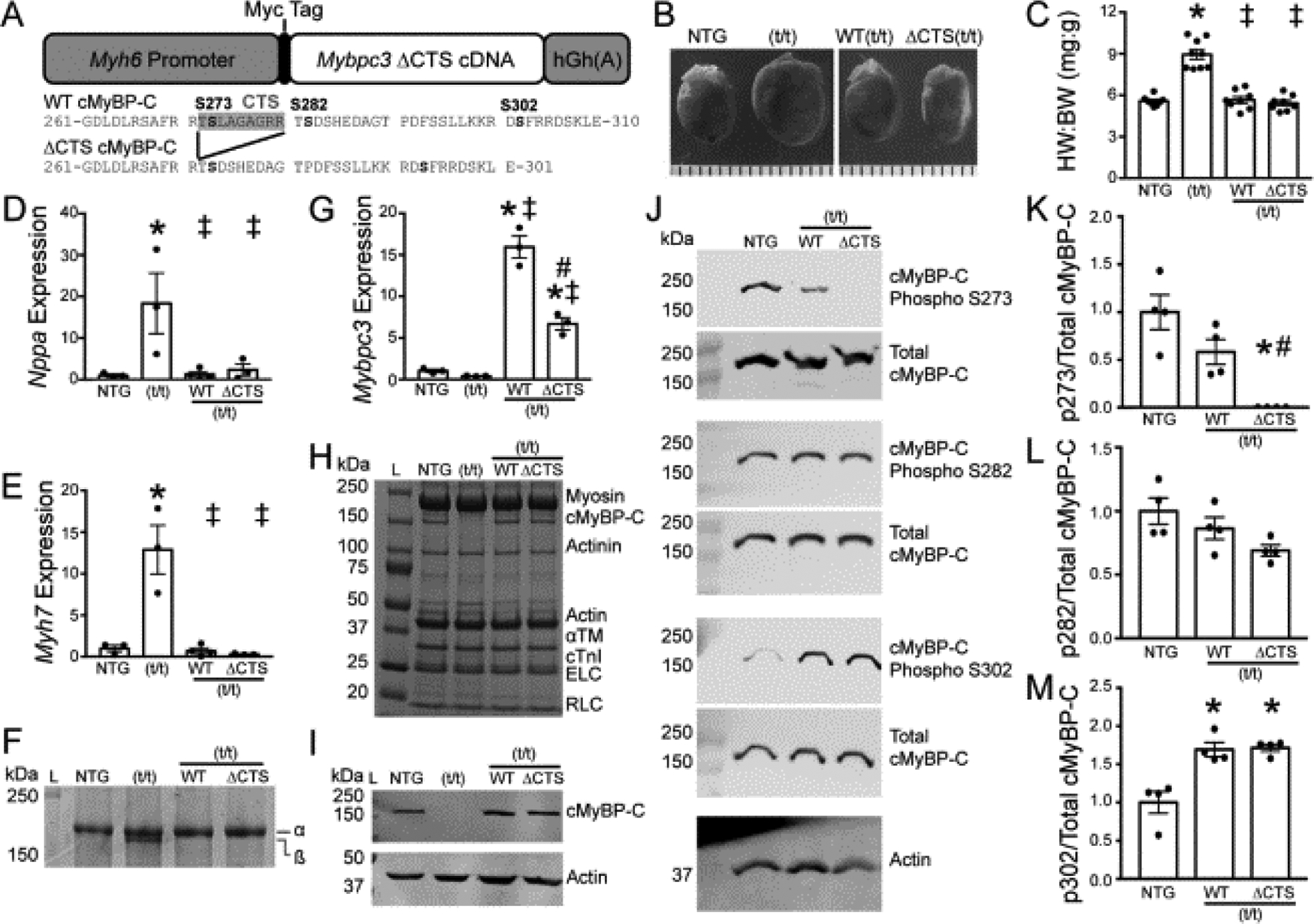
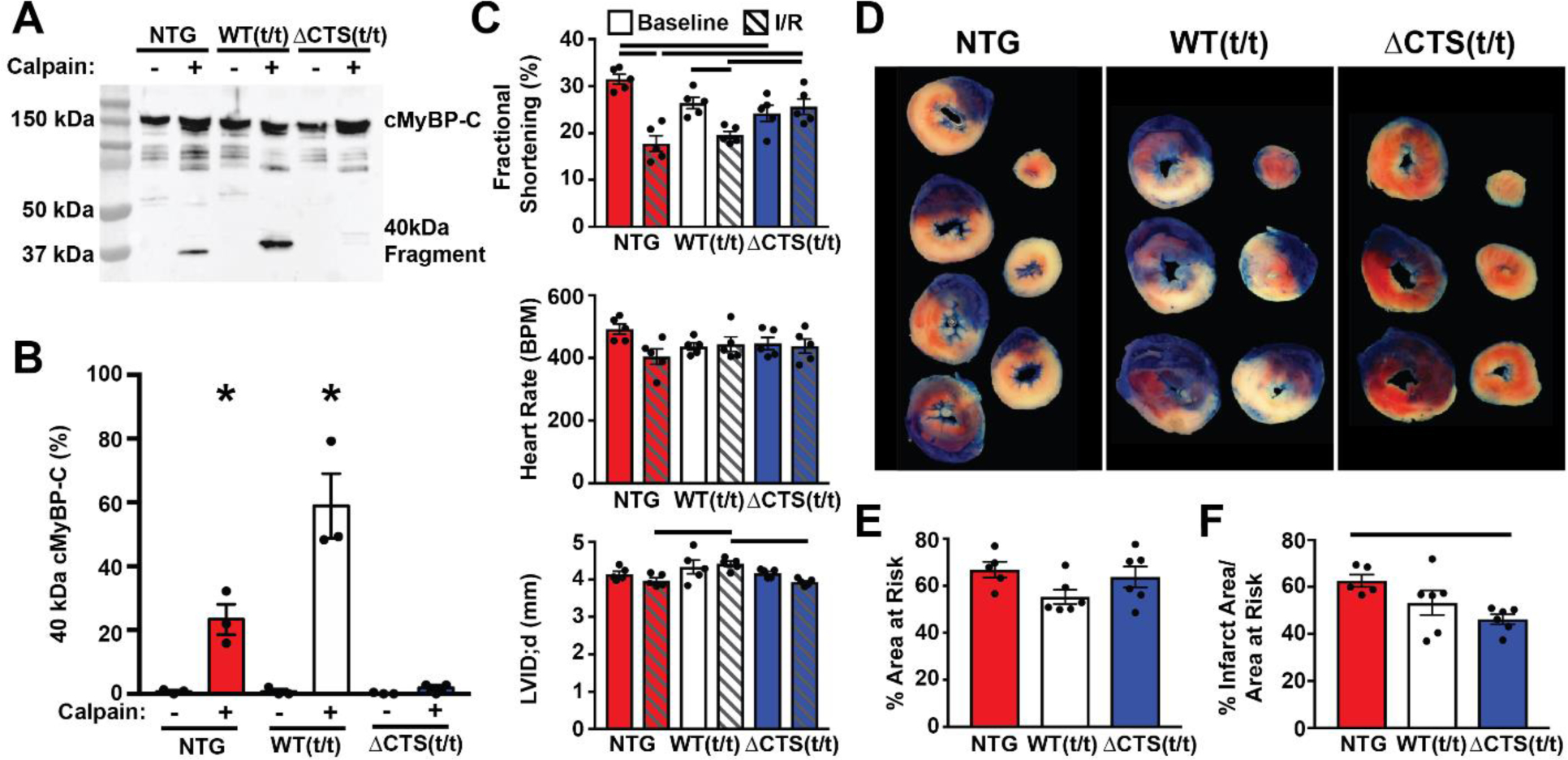
Cardiac myosin binding protein-C (cMyBP-C) phosphorylation is essential for normal heart function and protects the heart from ischemia-reperfusion (I/R) injury. It is known that protein kinase-A (PKA)-mediated phosphorylation of cMyBP-C prevents I/R-dependent proteolysis, whereas dephosphorylation of cMyBP-C at PKA sites correlates with its degradation. While sites on cMyBP-C associated with phosphorylation and proteolysis co-localize, the mechanisms that link cMyBP-C phosphorylation and proteolysis during cardioprotection are not well understood. Therefore, we aimed to determine if abrogation of cMyBP-C proteolysis in association with calpain, a calcium-activated protease, confers cardioprotection during I/R injury. Calpain is activated in both human ischemic heart samples and ischemic mouse myocardium where cMyBP-C is dephosphorylated and undergoes proteolysis. Moreover, cMyBP-C is a substrate for calpain proteolysis and cleaved by calpain at residues 272-TSLAGAGRR-280, a domain termed as the calpain-target site (CTS). Cardiac-specific transgenic (Tg) mice in which the CTS motif was ablated were bred into a cMyBP-C null background. These Tg mice were conclusively shown to possess a normal basal structure and function by analysis of histology, electron microscopy, immunofluorescence microscopy, Q-space MRI of tissue architecture, echocardiography, and hemodynamics. However, the genetic ablation of the CTS motif conferred resistance to calpain-mediated proteolysis of cMyBP-C. Following I/R injury, the loss of the CTS reduced infarct size compared to non-transgenic controls. Collectively, these findings demonstrate the physiological significance of calpain-targeted cMyBP-C proteolysis and provide a rationale for studying inhibition of calpain-mediated proteolysis of cMyBP-C as a therapeutic target for cardioprotection.
Keywords: Calpain; Cardioprotection; Ischemia-reperfusion injury; MYBPC3; cMyBP-C.
Copyright © 2019 The Authors. Published by Elsevier Ltd.. All rights reserved.
Figures


Similar articles
-
Cardiac ischemic preconditioning promotes cMyBP-C phosphorylation by inhibiting the calpain-mediated proteolysis.Exp Cell Res. 2023 Dec 15;433(2):113859. doi: 10.1016/j.yexcr.2023.113859. Epub 2023 Nov 22. Exp Cell Res. 2023. PMID: 38000772
-
Receptor-independent cardiac protein kinase Calpha activation by calpain-mediated truncation of regulatory domains.Circ Res. 2010 Oct 1;107(7):903-12. doi: 10.1161/CIRCRESAHA.110.220772. Epub 2010 Aug 5. Circ Res. 2010. PMID: 20689063 Free PMC article.
-
Cardiac myosin binding protein-C phosphorylation in a {beta}-myosin heavy chain background.Circulation. 2009 Mar 10;119(9):1253-62. doi: 10.1161/CIRCULATIONAHA.108.798983. Epub 2009 Feb 23. Circulation. 2009. PMID: 19237661 Free PMC article.
-
Phosphorylation and function of cardiac myosin binding protein-C in health and disease.J Mol Cell Cardiol. 2010 May;48(5):866-75. doi: 10.1016/j.yjmcc.2009.11.014. Epub 2009 Dec 3. J Mol Cell Cardiol. 2010. PMID: 19962384 Free PMC article. Review.
-
Surviving the infarct: A profile of cardiac myosin binding protein-C pathogenicity, diagnostic utility, and proteomics in the ischemic myocardium.Proteomics Clin Appl. 2014 Aug;8(7-8):569-77. doi: 10.1002/prca.201400011. Epub 2014 Jul 14. Proteomics Clin Appl. 2014. PMID: 24888514 Free PMC article. Review.
Cited by
-
Roles of cMyBP-C phosphorylation on cardiac contractile dysfunction in db/db mice.J Mol Cell Cardiol Plus. 2024 Jun;8:100075. doi: 10.1016/j.jmccpl.2024.100075. Epub 2024 Apr 4. J Mol Cell Cardiol Plus. 2024. PMID: 38957358 Free PMC article.
-
Phosphorylation of cardiac myosin-binding protein-C contributes to calcium homeostasis.J Biol Chem. 2020 Aug 7;295(32):11275-11291. doi: 10.1074/jbc.RA120.013296. Epub 2020 Jun 18. J Biol Chem. 2020. PMID: 32554466 Free PMC article.
-
Myofilament Alterations Associated with Human R14del-Phospholamban Cardiomyopathy.Int J Mol Sci. 2023 Jan 31;24(3):2675. doi: 10.3390/ijms24032675. Int J Mol Sci. 2023. PMID: 36768995 Free PMC article.
-
Under construction: The dynamic assembly, maintenance, and degradation of the cardiac sarcomere.J Mol Cell Cardiol. 2020 Nov;148:89-102. doi: 10.1016/j.yjmcc.2020.08.018. Epub 2020 Sep 10. J Mol Cell Cardiol. 2020. PMID: 32920010 Free PMC article. Review.
-
The roles of intracellular proteolysis in cardiac ischemia-reperfusion injury.Basic Res Cardiol. 2023 Sep 28;118(1):38. doi: 10.1007/s00395-023-01007-z. Basic Res Cardiol. 2023. PMID: 37768438 Review.
References
-
- Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, E.S.C.A.A.H.A.W.H.F.T.F.f.U.D.o.M.I. Joint, C. Authors/Task Force Members, Thygesen K, Alpert JS, White HD, Biomarker S, Jaffe AS, Katus HA, Apple FS, Lindahl B, Morrow DA, E.C.G. Subcommittee, Chaitman BR, Clemmensen PM, Johanson P, Hod H, Imaging S, Underwood R, Bax JJ, Bonow JJ, Pinto F, Gibbons RJ, Classification S, Fox KA, Atar D, Newby LK, Galvani M, Hamm CW, Intervention S, Uretsky BF, Steg PG, Wijns W, Bassand JP, Menasche P, Ravkilde J, Trials, S. Registries, Ohman EM, Antman EM, Wallentin LC, Armstrong PW, Simoons ML, Trials, S. Registries, Januzzi JL, Nieminen MS, Gheorghiade M, Filippatos G, Trials, S. Registries, Luepker RV, Fortmann SP, Rosamond WD, Levy D, Wood D, Trials, S. Registries, Smith SC, Hu D, Lopez-Sendon JL, Robertson RM, Weaver D, Tendera M, Bove AA, Parkhomenko AN, Vasilieva EJ, Mendis S, E.S.C.C.f.P. Guidelines, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, Document R, Morais J, Aguiar C, Almahmeed W, Arnar DO, Barili F, Bloch KD, Bolger AF, Botker HE, Bozkurt B, Bugiardini R, Cannon C, Lemos J. de, Eberli FR, Escobar E, Hlatky M, James S, Kern KB, Moliterno DJ, Mueller C, Neskovic AN, Pieske BM, Schulman SP, Storey RF, Taubert KA, Vranckx P, Wagner DR , Third universal definition of myocardial infarction, J Am Coll Cardiol 60(16) (2012) 1581–98. - PubMed
-
- Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American E Heart Association Council on, C. Prevention Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association, Circulation 137(12) (2018) e67–e492. - PubMed
-
- van den Borne SW, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J, Myocardial remodeling after infarction: the role of myofibroblasts, Nat Rev Cardiol 7(1) (2010) 30–7. - PubMed
-
- Daskalopoulos EP, Janssen BJ, Blankesteijn WM, Myofibroblasts in the infarct area: concepts and challenges, Microsc Microanal 18(1) (2012) 35–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 DC011528/DC/NIDCD NIH HHS/United States
- RG/11/21/29335/BHF_/British Heart Foundation/United Kingdom
- R01 HL131517/HL/NHLBI NIH HHS/United States
- R01 AA024769/AA/NIAAA NIH HHS/United States
- F32 HL131304/HL/NHLBI NIH HHS/United States
- R01 HL113640/HL/NHLBI NIH HHS/United States
- K02 HL114749/HL/NHLBI NIH HHS/United States
- R01 HL136389/HL/NHLBI NIH HHS/United States
- R01 HL130356/HL/NHLBI NIH HHS/United States
- R01 HL146744/HL/NHLBI NIH HHS/United States
- R01 HL168728/HL/NHLBI NIH HHS/United States
- R01 HL105826/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
