

Molecular epidemiology, evolution and phylogeny of SARS coronavirus
- PMID: 30844511
- PMCID: PMC7106202
- DOI: 10.1016/j.meegid.2019.03.001
Molecular epidemiology, evolution and phylogeny of SARS coronavirus
Abstract
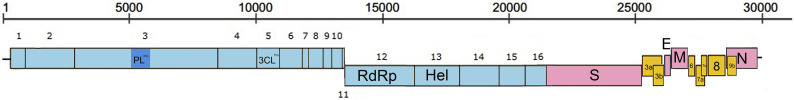
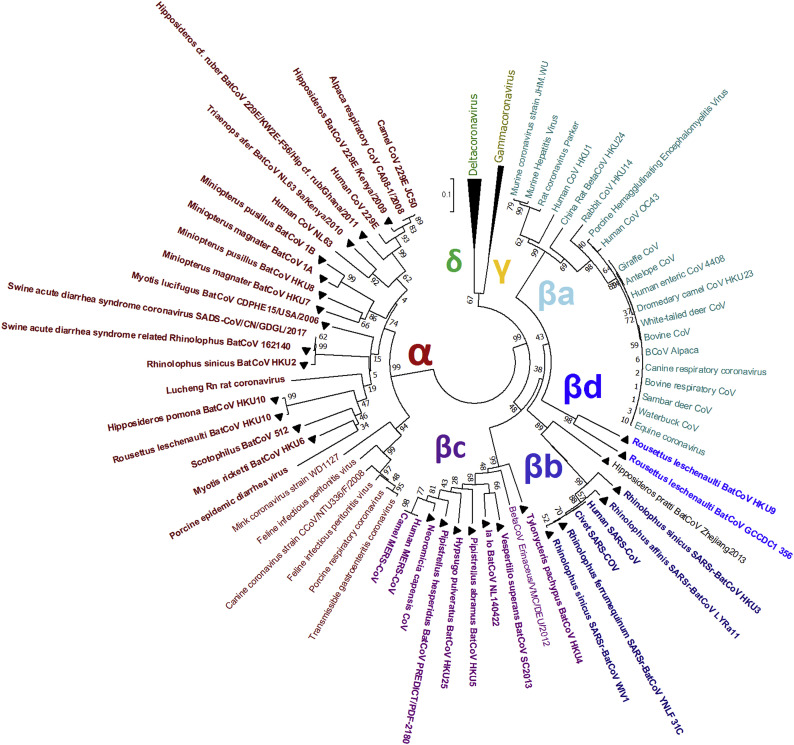
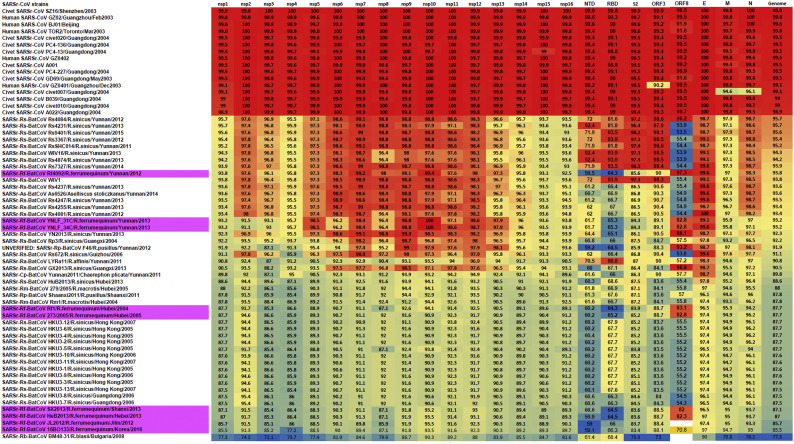
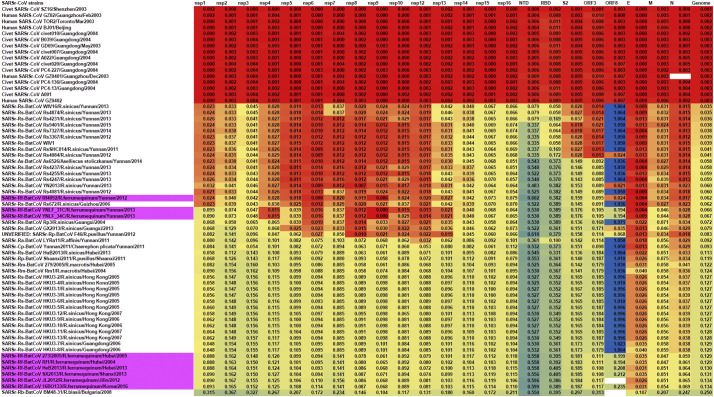
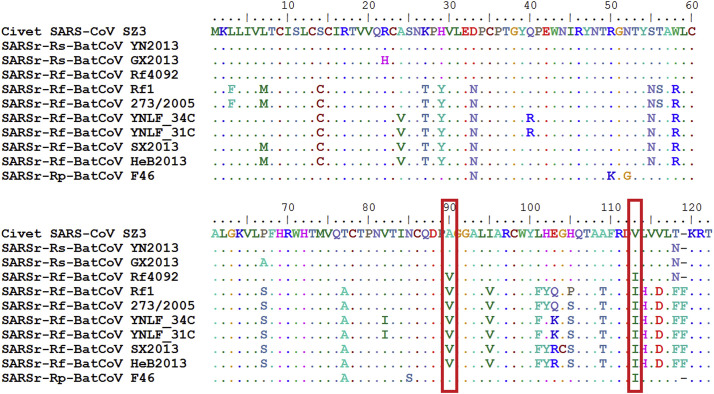
Shortly after its emergence in southern China in 2002/2003, Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) was confirmed to be the cause of SARS. Subsequently, SARS-related CoVs (SARSr-CoVs) were found in palm civets from live animal markets in Guangdong and in various horseshoe bat species, which were believed to be the ultimate reservoir of SARSr-CoV. Till November 2018, 339 SARSr-CoV genomes have been sequenced, including 274 from human, 18 from civets and 47 from bats [mostly from Chinese horseshoe bats (Rhinolophus sinicus), n = 30; and greater horseshoe bats (Rhinolophus ferrumequinum), n = 9]. The human SARS-CoVs and civet SARSr-CoVs were collected in 2003/2004, while bat SARSr-CoVs were continuously isolated in the past 13 years even after the cessation of the SARS epidemic. SARSr-CoVs belong to the subgenus Sarbecovirus (previously lineage B) of genus Betacoronavirus and occupy a unique phylogenetic position. Overall, it is observed that the SARSr-CoV genomes from bats in Yunnan province of China possess the highest nucleotide identity to those from civets. It is evident from both multiple alignment and phylogenetic analyses that some genes of a particular SARSr-CoV from bats may possess higher while other genes possess much lower nucleotide identity to the corresponding genes of SARSr-CoV from human/civets, resulting in the shift of phylogenetic position in different phylogenetic trees. Our current model on the origin of SARS is that the human SARS-CoV that caused the epidemic in 2002/2003 was probably a result of multiple recombination events from a number of SARSr-CoV ancestors in different horseshoe bat species.
Keywords: Evolution; Molecular epidemiology; Phylogeny; SARS coronavirus.
Copyright © 2019. Published by Elsevier B.V.
Figures


Similar articles
-
Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination.J Virol. 2015 Oct;89(20):10532-47. doi: 10.1128/JVI.01048-15. Epub 2015 Aug 12. J Virol. 2015. PMID: 26269185 Free PMC article.
-
Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China.mBio. 2022 Jun 28;13(3):e0046322. doi: 10.1128/mbio.00463-22. Epub 2022 Apr 25. mBio. 2022. PMID: 35467426 Free PMC article.
-
Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events.J Virol. 2010 Mar;84(6):2808-19. doi: 10.1128/JVI.02219-09. Epub 2010 Jan 13. J Virol. 2010. PMID: 20071579 Free PMC article.
-
Geographical structure of bat SARS-related coronaviruses.Infect Genet Evol. 2019 Apr;69:224-229. doi: 10.1016/j.meegid.2019.02.001. Epub 2019 Feb 6. Infect Genet Evol. 2019. PMID: 30735813 Free PMC article. Review.
-
A review of studies on animal reservoirs of the SARS coronavirus.Virus Res. 2008 Apr;133(1):74-87. doi: 10.1016/j.virusres.2007.03.012. Epub 2007 Apr 23. Virus Res. 2008. PMID: 17451830 Free PMC article. Review.
Cited by
-
Surveillance strategies for SARS-CoV-2 infections through one health approach.Heliyon. 2024 Aug 30;10(17):e37128. doi: 10.1016/j.heliyon.2024.e37128. eCollection 2024 Sep 15. Heliyon. 2024. PMID: 39286214 Free PMC article. Review.
-
Rapid antigen detection of severe acute respiratory syndrome coronavirus-2 in stray cats: A cross-sectional study.Vet World. 2024 Jul;17(7):1611-1618. doi: 10.14202/vetworld.2024.1611-1618. Epub 2024 Jul 26. Vet World. 2024. PMID: 39185047 Free PMC article.
-
Exploring Cannabinoids as Potential Inhibitors of SARS-CoV-2 Papain-like Protease: Insights from Computational Analysis and Molecular Dynamics Simulations.Viruses. 2024 May 30;16(6):878. doi: 10.3390/v16060878. Viruses. 2024. PMID: 38932170 Free PMC article.
-
A tale of endurance: bats, viruses and immune dynamics.Future Microbiol. 2024 Jun 12;19(9):841-856. doi: 10.2217/fmb-2023-0233. Epub 2024 Apr 22. Future Microbiol. 2024. PMID: 38648093 Review.
-
Bat-associated microbes: Opportunities and perils, an overview.Heliyon. 2023 Nov 18;9(12):e22351. doi: 10.1016/j.heliyon.2023.e22351. eCollection 2023 Dec. Heliyon. 2023. PMID: 38125540 Free PMC article. Review.
References
-
- de Groot R.J., Baker S.C., Baric R., Enjuanes L., Gorbalenya A., Holmes K.V., Perlman S., Poon L., Rottier P.J., Talbot P.J., Woo P.C., Ziebuhr J. Virus Taxonomy, Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses. International Union of Microbiological Societies, Virology Division; 2011. Coronaviridae; pp. 806–828.
-
- Drexler J.F., Gloza-Rausch F., Glende J., Corman V.M., Muth D., Goettsche M., Seebens A., Niedrig M., Pfefferle S., Yordanov S., Zhelyazkov L., Hermanns U., Vallo P., Lukashev A., Muller M.A., Deng H., Herrler G., Drosten C. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010;84:11336–11349. - PMC - PubMed
-
- Ge X.Y., Li J.L., Yang X.L., Chmura A.A., Zhu G., Epstein J.H., Mazet J.K., Hu B., Zhang W., Peng C., Zhang Y.J., Luo C.M., Tan B., Wang N., Zhu Y., Crameri G., Zhang S.Y., Wang L.F., Daszak P., Shi Z.L. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013;503:535–538. - PMC - PubMed
-
- Guan Y., Zheng B.J., He Y.Q., Liu X.L., Zhuang Z.X., Cheung C.L., Luo S.W., Li P.H., Zhang L.J., Guan Y.J., Butt K.M., Wong K.L., Chan K.W., Lim W., Shortridge K.F., Yuen K.Y., Peiris J.S., Poon L.L. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
