

Statins Perturb G βγ Signaling and Cell Behavior in a G γ Subtype Dependent Manner
- PMID: 30765461
- PMCID: PMC6402420
- DOI: 10.1124/mol.118.114710
Statins Perturb G βγ Signaling and Cell Behavior in a G γ Subtype Dependent Manner
Abstract
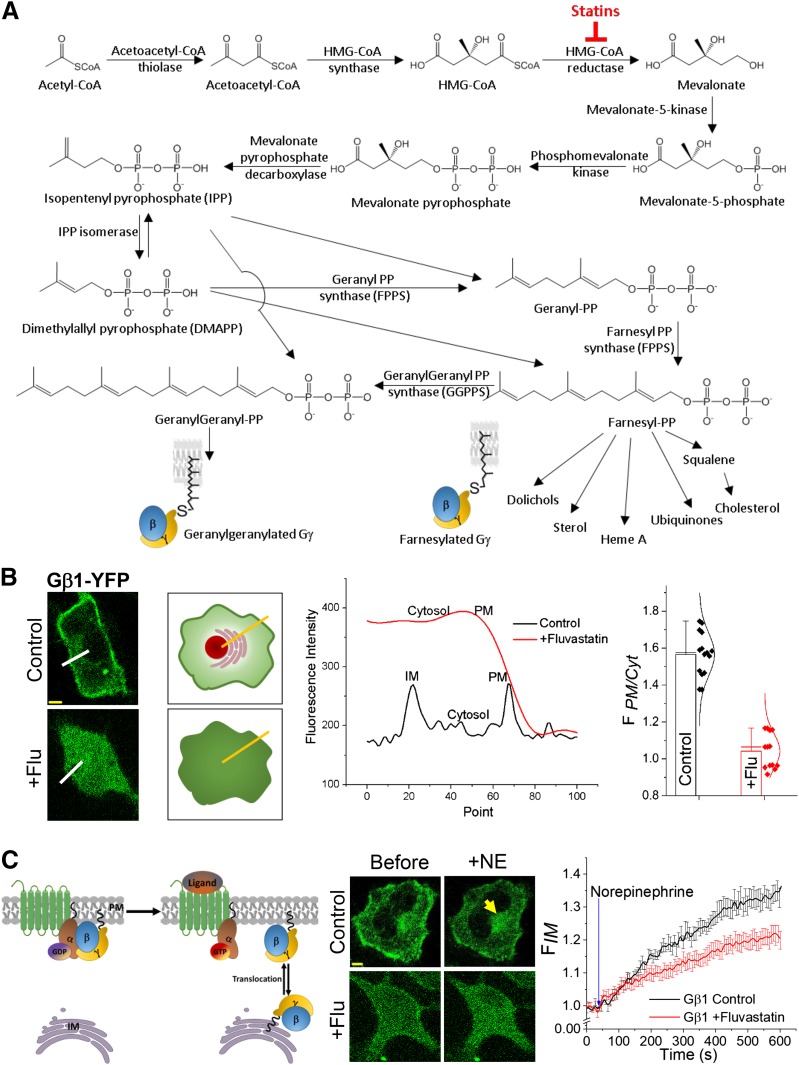
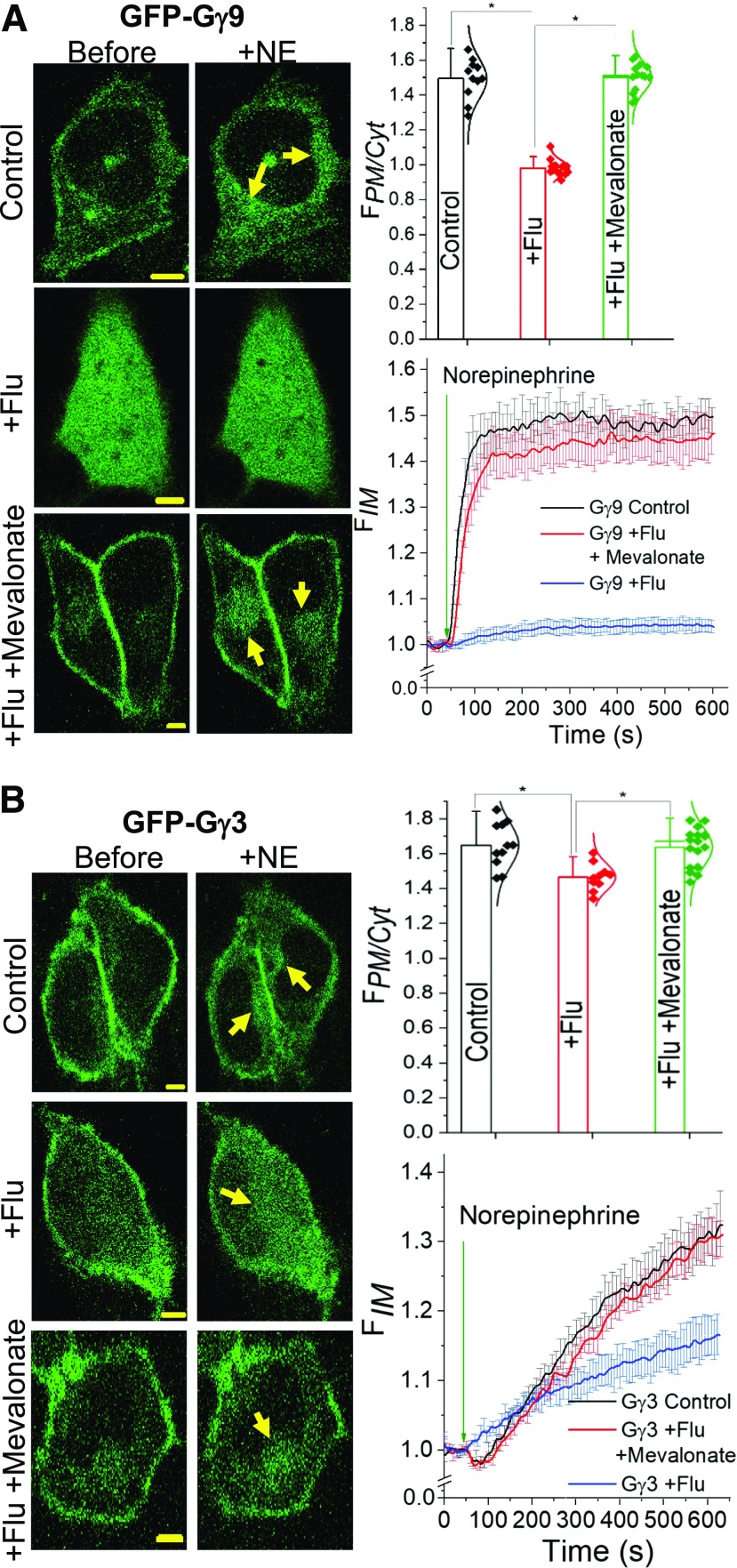
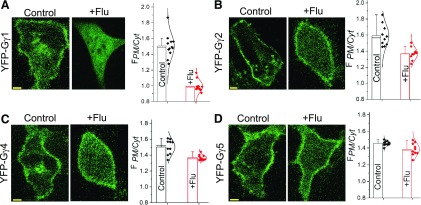
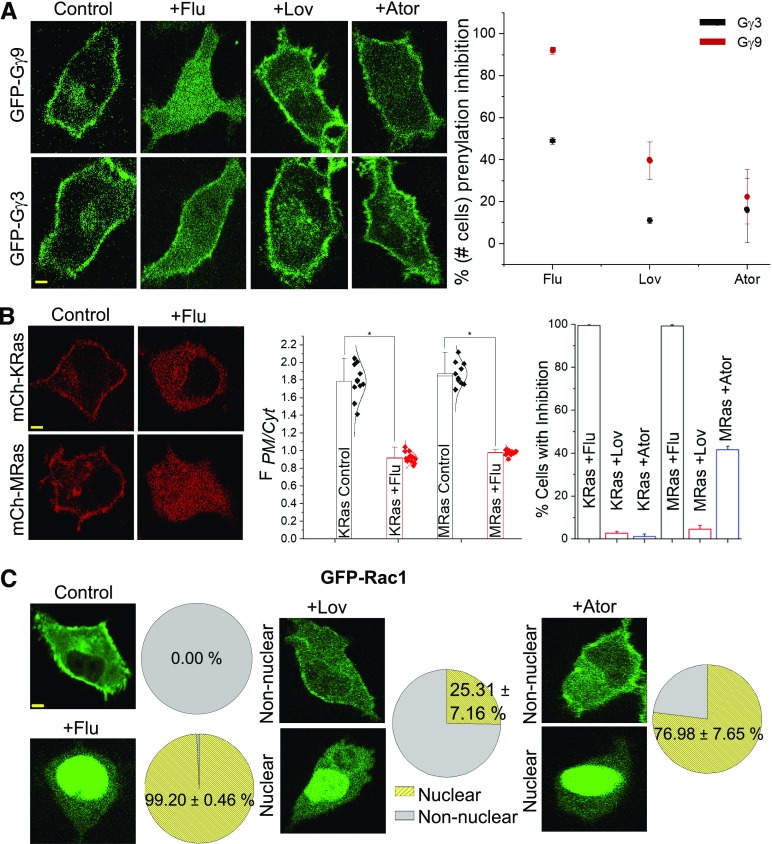
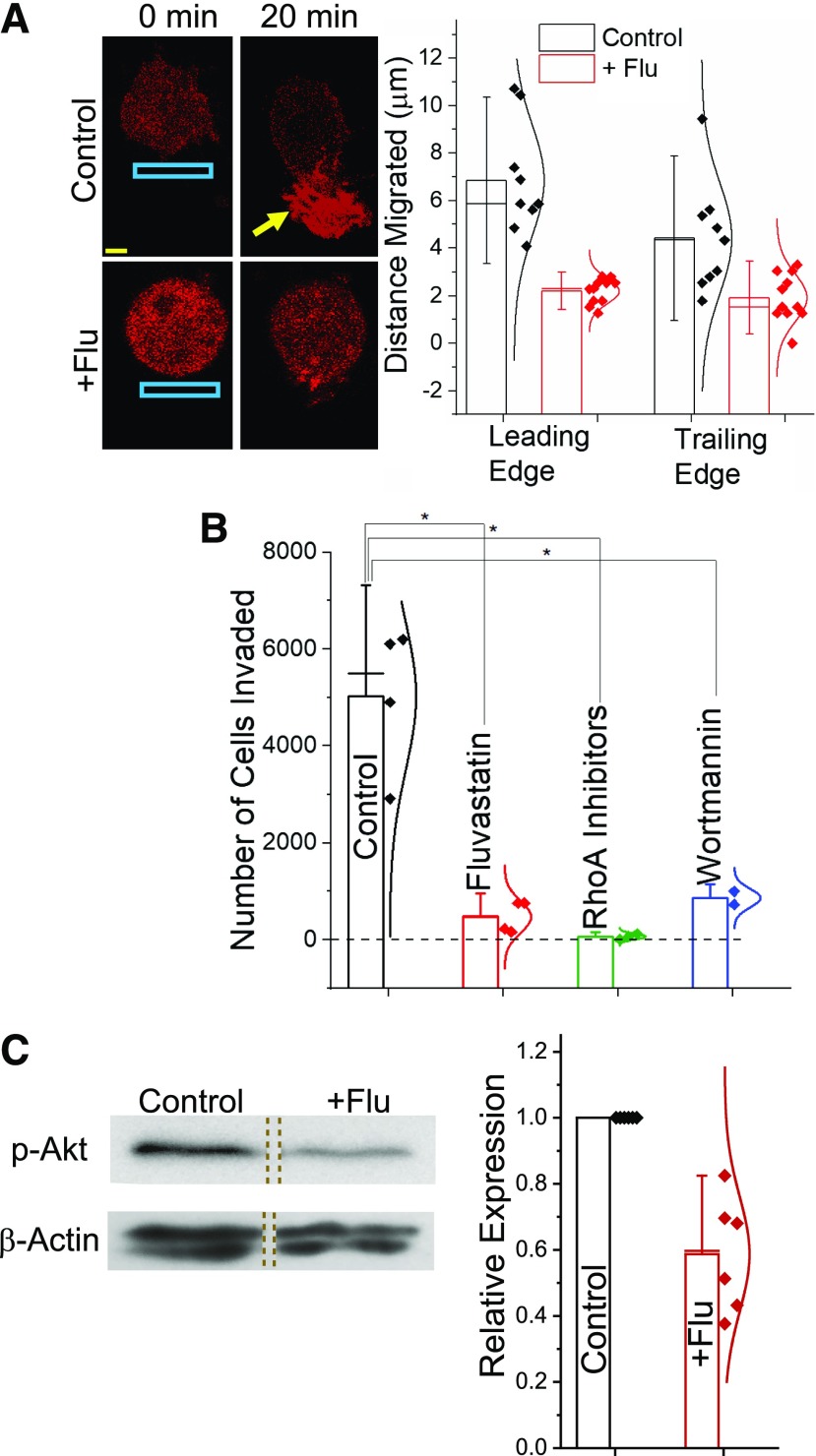
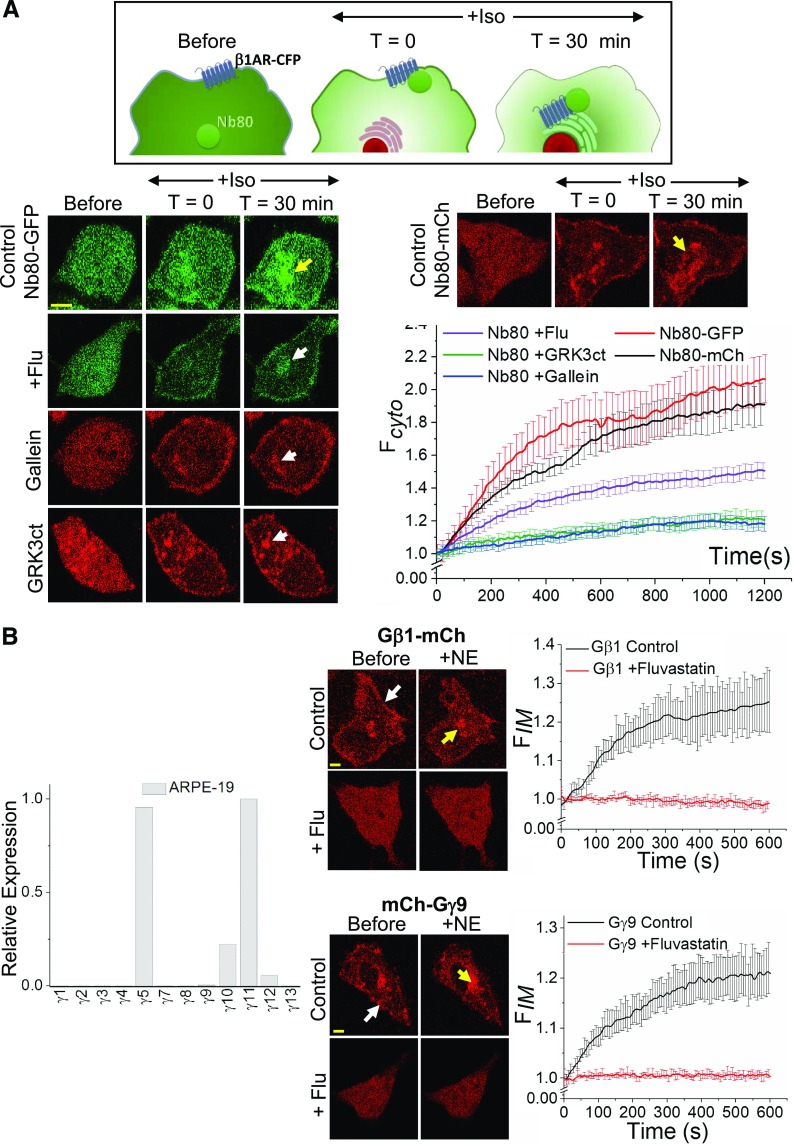
Guanine nucleotide-binding proteins (G proteins) facilitate the transduction of external signals to the cell interior, regulate most eukaryotic signaling, and thus have become crucial disease drivers. G proteins largely function at the inner leaflet of the plasma membrane (PM) using covalently attached lipid anchors. Both small monomeric and heterotrimeric G proteins are primarily prenylated, either with a 15-carbon farnesyl or a 20-carbon geranylgeranyl polyunsaturated lipid. The mevalonate [3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase] pathway synthesizes lipids for G-protein prenylation. It is also the source of the precursor lipids for many biomolecules, including cholesterol. Consequently, the rate-limiting enzymes of the mevalonate pathway are major targets for cholesterol-lowering medications and anticancer drug development. Although prenylated G protein γ (Gγ) is essential for G protein-coupled receptor (GPCR)-mediated signaling, how mevalonate pathway inhibitors, statins, influence subcellular distribution of Gβγ dimer and Gαβγ heterotrimer, as well as their signaling upon GPCR activation, is poorly understood. The present study shows that clinically used statins not only significantly disrupt PM localization of Gβγ but also perturb GPCR-G protein signaling and associated cell behaviors. The results also demonstrate that the efficiency of prenylation inhibition by statins is Gγ subtype-dependent and is more effective toward farnesylated Gγ types. Since Gγ is required for Gβγ signaling and shows a cell- and tissue-specific subtype distribution, the present study can help understand the mechanisms underlying clinical outcomes of statin use in patients. This work also reveals the potential of statins as clinically usable drugs to control selected GPCR-G protein signaling.
Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics.
Figures

Similar articles
-
Gγ identity dictates efficacy of Gβγ signaling and macrophage migration.J Biol Chem. 2018 Feb 23;293(8):2974-2989. doi: 10.1074/jbc.RA117.000872. Epub 2018 Jan 9. J Biol Chem. 2018. PMID: 29317505 Free PMC article.
-
CaaX-motif-adjacent residues influence G protein gamma (Gγ) prenylation under suboptimal conditions.J Biol Chem. 2023 Nov;299(11):105269. doi: 10.1016/j.jbc.2023.105269. Epub 2023 Sep 20. J Biol Chem. 2023. PMID: 37739036 Free PMC article.
-
A short C-terminal peptide in Gγ regulates Gβγ signaling efficacy.Mol Biol Cell. 2021 Aug 1;32(16):1446-1458. doi: 10.1091/mbc.E20-11-0750. Epub 2021 Jun 9. Mol Biol Cell. 2021. PMID: 34106735 Free PMC article.
-
Subtype-dependent regulation of Gβγ signalling.Cell Signal. 2021 Jun;82:109947. doi: 10.1016/j.cellsig.2021.109947. Epub 2021 Feb 11. Cell Signal. 2021. PMID: 33582184 Free PMC article. Review.
-
Regulation of G Protein βγ Signaling.Int Rev Cell Mol Biol. 2018;339:133-191. doi: 10.1016/bs.ircmb.2018.02.008. Epub 2018 Mar 28. Int Rev Cell Mol Biol. 2018. PMID: 29776603 Review.
Cited by
-
Optical approaches for single-cell and subcellular analysis of GPCR-G protein signaling.Anal Bioanal Chem. 2019 Jul;411(19):4481-4508. doi: 10.1007/s00216-019-01774-6. Epub 2019 Mar 30. Anal Bioanal Chem. 2019. PMID: 30927013 Free PMC article. Review.
-
Synthesis of the 6-Substituted Imidazo[1,2-a]Pyridine-3-yl-2- Phosphonopropionic Acids as Potential Inhibitors of Rab Geranylgeranyl Transferase.Front Chem. 2021 Jan 6;8:596162. doi: 10.3389/fchem.2020.596162. eCollection 2020. Front Chem. 2021. PMID: 33490034 Free PMC article.
-
CaaX-motif adjacent residues control G protein prenylation under suboptimal conditions.bioRxiv [Preprint]. 2023 Jul 4:2023.07.04.547731. doi: 10.1101/2023.07.04.547731. bioRxiv. 2023. Update in: J Biol Chem. 2023 Nov;299(11):105269. doi: 10.1016/j.jbc.2023.105269. PMID: 37461501 Free PMC article. Updated. Preprint.
-
Effects of Statins on Renin-Angiotensin System.J Cardiovasc Dev Dis. 2021 Jul 9;8(7):80. doi: 10.3390/jcdd8070080. J Cardiovasc Dev Dis. 2021. PMID: 34357323 Free PMC article. Review.
-
Dissociation of the G protein βγ from the Gq-PLCβ complex partially attenuates PIP2 hydrolysis.J Biol Chem. 2021 Jan-Jun;296:100702. doi: 10.1016/j.jbc.2021.100702. Epub 2021 Apr 24. J Biol Chem. 2021. PMID: 33901492 Free PMC article.
References
-
- Alberts AW. (1988) Discovery, biochemistry and biology of lovastatin. Am J Cardiol 62:10J–15J. - PubMed
-
- Bathgate-Siryk A, Dabul S, Pandya K, Walklett K, Rengo G, Cannavo A, De Lucia C, Liccardo D, Gao E, Leosco D, et al. (2014) Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension 63(2):404–412. - PMC - PubMed
-
- Braselmann S, Palmer TM, Cook SJ. (1997) Signalling enzymes: bursting with potential. Curr Biol 7:R470–R473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials