

Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program
- PMID: 30686120
- PMCID: PMC6459716
- DOI: 10.1161/CIRCRESAHA.118.314438
Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program
Abstract
Rationale: The heart contains abundant interstitial and perivascular fibroblasts. Traditional views suggest that, under conditions of mechanical stress, cytokines, growth factors, and neurohumoral mediators stimulate fibroblast activation, inducing ECM (extracellular matrix) protein synthesis and promoting fibrosis and diastolic dysfunction. Members of the TGF (transforming growth factor)-β family are upregulated and activated in the remodeling myocardium and modulate phenotype and function of all myocardial cell types through activation of intracellular effector molecules, the Smads (small mothers against decapentaplegic), and through Smad-independent pathways.
Objectives: To examine the role of fibroblast-specific TGF-β/Smad3 signaling in the remodeling pressure-overloaded myocardium.
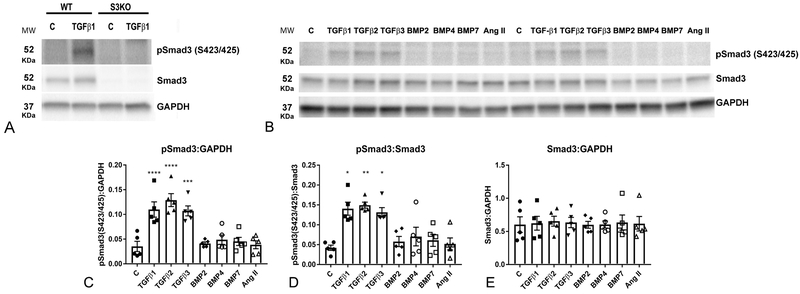
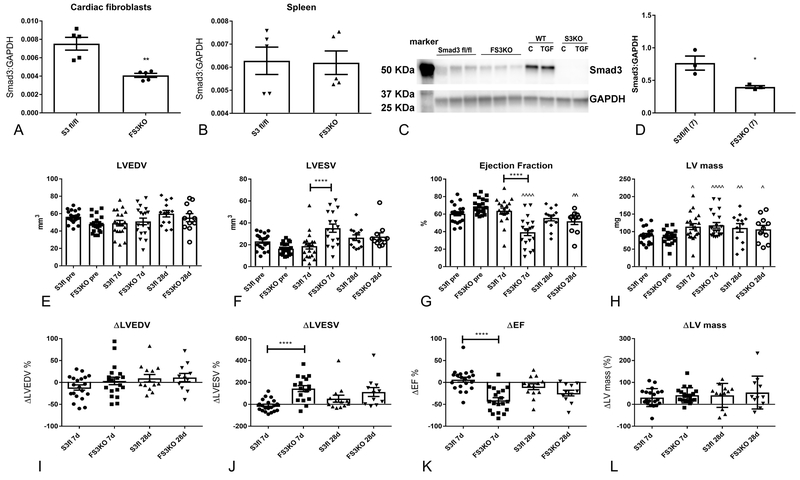
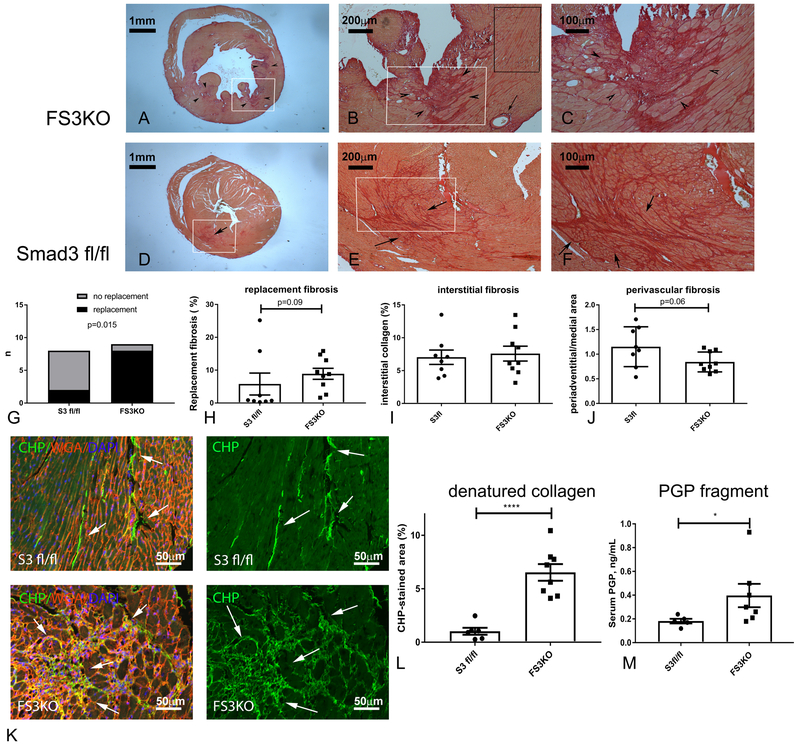
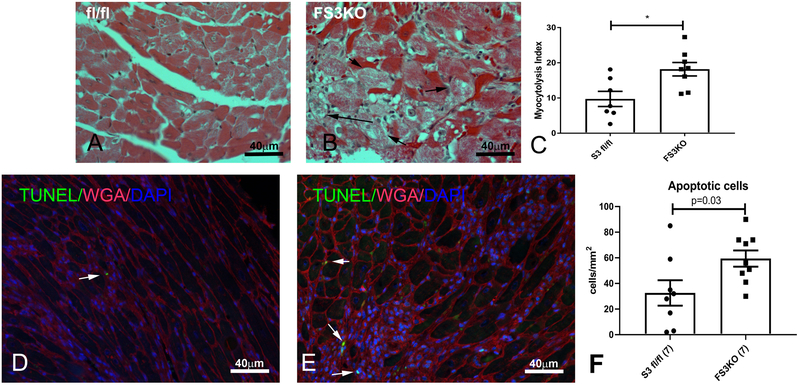
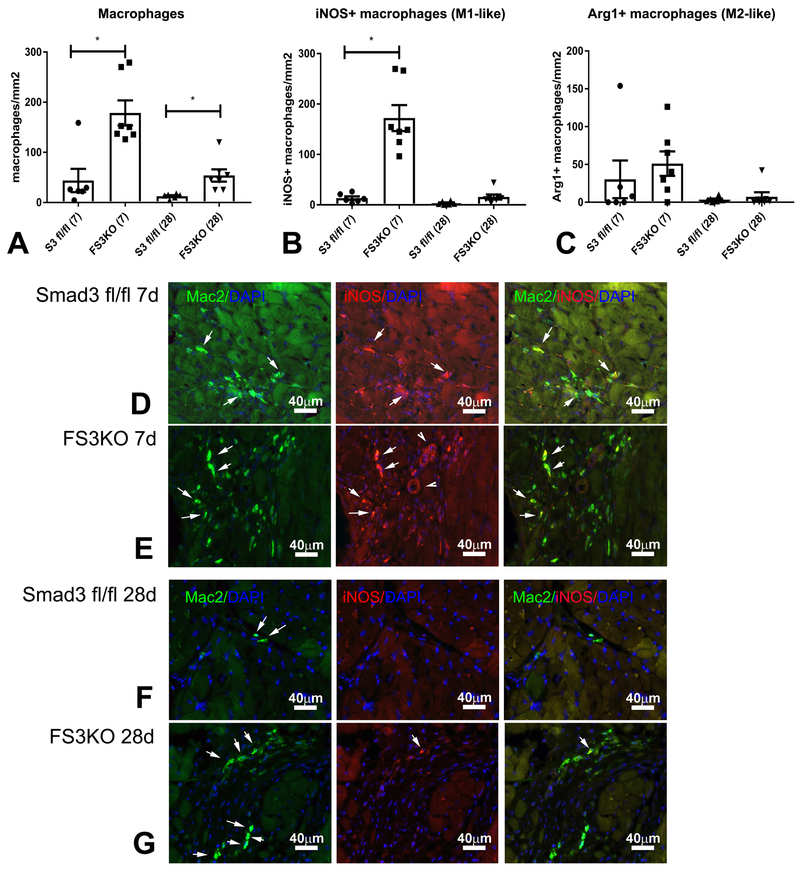
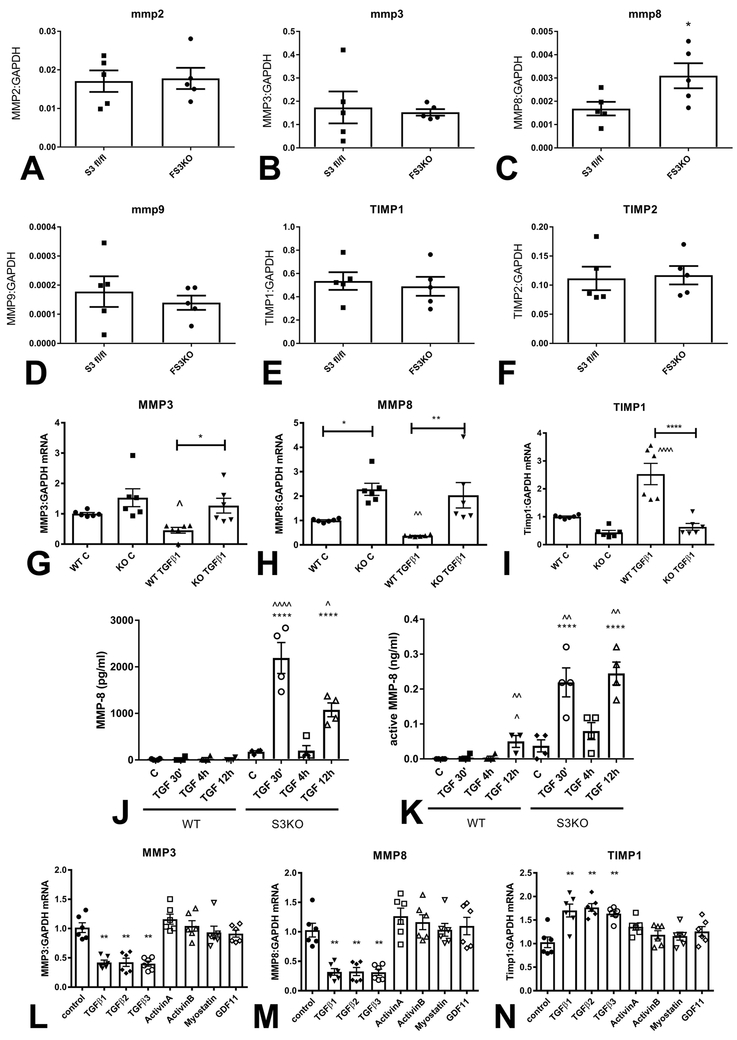
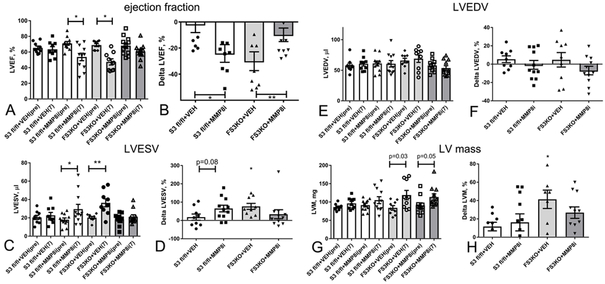
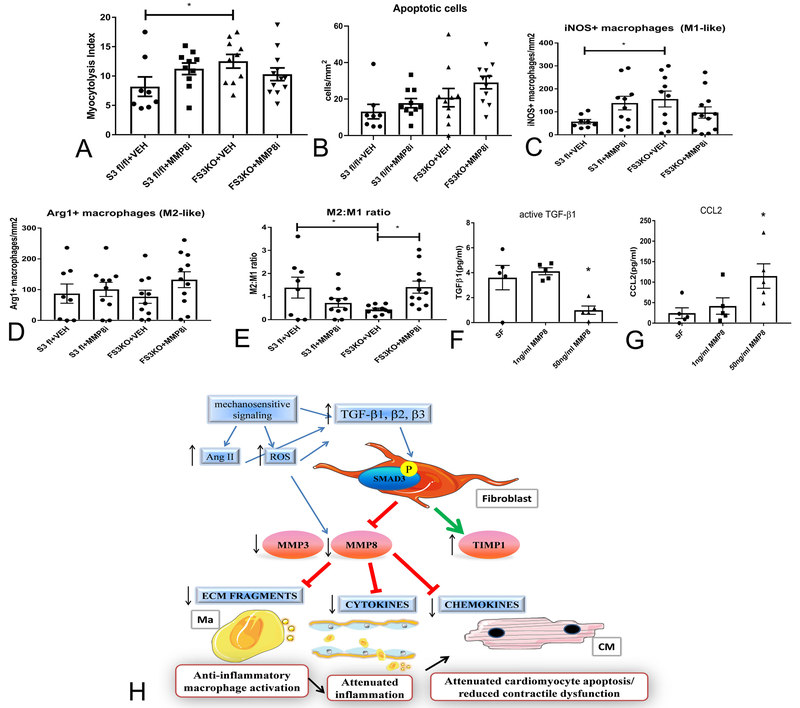
Methods and results: We examined the effects of cell-specific Smad3 loss in activated periostin-expressing myofibroblasts using a mouse model of cardiac pressure overload, induced through transverse aortic constriction. Surprisingly, FS3KO (myofibroblast-specific Smad3 knockout) mice exhibited accelerated systolic dysfunction after pressure overload, evidenced by an early 40% reduction in ejection fraction after 7 days of transverse aortic constriction. Accelerated systolic dysfunction in pressure-overloaded FS3KO mice was associated with accentuated matrix degradation and generation of collagen-derived matrikines, accompanied by cardiomyocyte myofibrillar loss and apoptosis, and by enhanced macrophage-driven inflammation. In vitro, TGF-β1, TGF-β2, and TGF-β3 stimulated a Smad3-dependent matrix-preserving phenotype in cardiac fibroblasts, suppressing MMP (matrix metalloproteinase)-3 and MMP-8 synthesis and inducing TIMP (tissue inhibitor of metalloproteinases)-1. In vivo, administration of an MMP-8 inhibitor attenuated early systolic dysfunction in pressure-overloaded FS3KO mice, suggesting that the protective effects of activated cardiac myofibroblasts in the pressure-overloaded myocardium are, at least in part, because of suppression of MMPs and activation of a matrix-preserving program. MMP-8 stimulation induces a proinflammatory phenotype in isolated macrophages.
Conclusions: In the pressure-overloaded myocardium, TGF-β/Smad3-activated cardiac fibroblasts play an important protective role, preserving the ECM network, suppressing macrophage-driven inflammation, and attenuating cardiomyocyte injury. The protective actions of the myofibroblasts are mediated, at least in part, through Smad-dependent suppression of matrix-degrading proteases.
Keywords: extracellular matrix; fibroblasts; inflammation; macrophages; matrix metalloproteinases.
Figures
Comment in
-
Maintaining Matrix Composure Under Stress.Circ Res. 2019 Apr 12;124(8):1149-1150. doi: 10.1161/CIRCRESAHA.119.314843. Circ Res. 2019. PMID: 30973810 No abstract available.
Similar articles
-
Fibroblast Smad7 Induction Protects the Remodeling Pressure-Overloaded Heart.Circ Res. 2024 Jul 19;135(3):453-469. doi: 10.1161/CIRCRESAHA.123.323360. Epub 2024 Jun 20. Circ Res. 2024. PMID: 38899461
-
Tissue transglutaminase induction in the pressure-overloaded myocardium regulates matrix remodelling.Cardiovasc Res. 2017 Jul 1;113(8):892-905. doi: 10.1093/cvr/cvx053. Cardiovasc Res. 2017. PMID: 28371893 Free PMC article.
-
Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart.J Mol Cell Cardiol. 2019 Jul;132:84-97. doi: 10.1016/j.yjmcc.2019.05.006. Epub 2019 May 11. J Mol Cell Cardiol. 2019. PMID: 31085202 Free PMC article.
-
Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities.Mol Aspects Med. 2019 Feb;65:70-99. doi: 10.1016/j.mam.2018.07.001. Epub 2018 Aug 2. Mol Aspects Med. 2019. PMID: 30056242 Review.
-
The role of Smad signaling cascades in cardiac fibrosis.Cell Signal. 2021 Jan;77:109826. doi: 10.1016/j.cellsig.2020.109826. Epub 2020 Nov 5. Cell Signal. 2021. PMID: 33160018 Free PMC article. Review.
Cited by
-
The heart under pressure: immune cells in fibrotic remodeling.Curr Opin Physiol. 2022 Feb;25:100484. doi: 10.1016/j.cophys.2022.100484. Epub 2022 Jan 22. Curr Opin Physiol. 2022. PMID: 35224321 Free PMC article.
-
Epigenetic Regulation of Fibroblasts and Crosstalk between Cardiomyocytes and Non-Myocyte Cells in Cardiac Fibrosis.Biomolecules. 2023 Sep 12;13(9):1382. doi: 10.3390/biom13091382. Biomolecules. 2023. PMID: 37759781 Free PMC article. Review.
-
Extracellular Matrix in Ischemic Heart Disease, Part 4/4: JACC Focus Seminar.J Am Coll Cardiol. 2020 May 5;75(17):2219-2235. doi: 10.1016/j.jacc.2020.03.020. J Am Coll Cardiol. 2020. PMID: 32354387 Free PMC article. Review.
-
Cardiac Fibrosis and Cardiac Fibroblast Lineage-Tracing: Recent Advances.Front Physiol. 2020 May 6;11:416. doi: 10.3389/fphys.2020.00416. eCollection 2020. Front Physiol. 2020. PMID: 32435205 Free PMC article. Review.
-
Transforming Growth Factor-β: A Multifunctional Regulator of Cancer Immunity.Cancers (Basel). 2020 Oct 23;12(11):3099. doi: 10.3390/cancers12113099. Cancers (Basel). 2020. PMID: 33114183 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
