

Protein Phosphatase 2A Regulates Cardiac Na+ Channels
- PMID: 30602331
- PMCID: PMC6395500
- DOI: 10.1161/CIRCRESAHA.118.314350
Protein Phosphatase 2A Regulates Cardiac Na+ Channels
Abstract
Rationale: Voltage-gated Na+ channel ( INa) function is critical for normal cardiac excitability. However, the Na+ channel late component ( INa,L) is directly associated with potentially fatal forms of congenital and acquired human arrhythmia. CaMKII (Ca2+/calmodulin-dependent kinase II) enhances INa,L in response to increased adrenergic tone. However, the pathways that negatively regulate the CaMKII/Nav1.5 axis are unknown and essential for the design of new therapies to regulate the pathogenic INa,L.
Objective: To define phosphatase pathways that regulate INa,L in vivo.
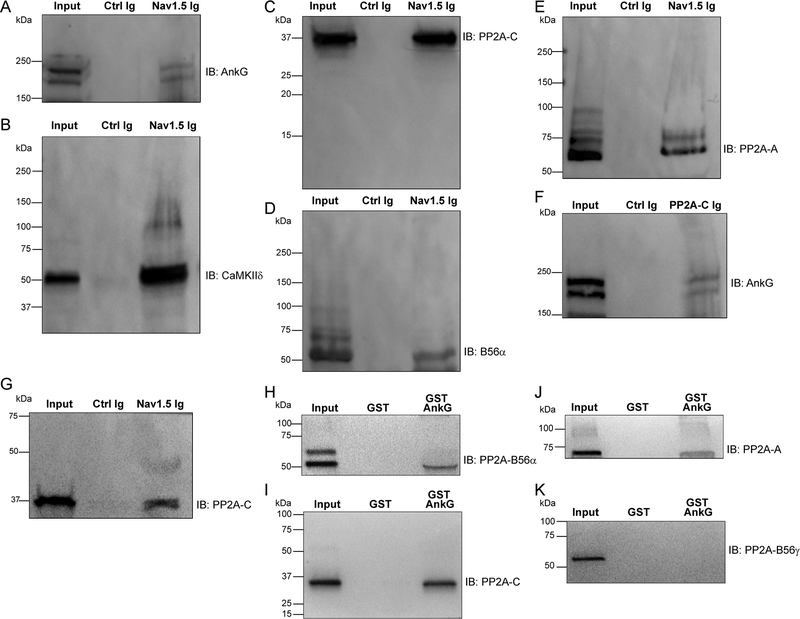
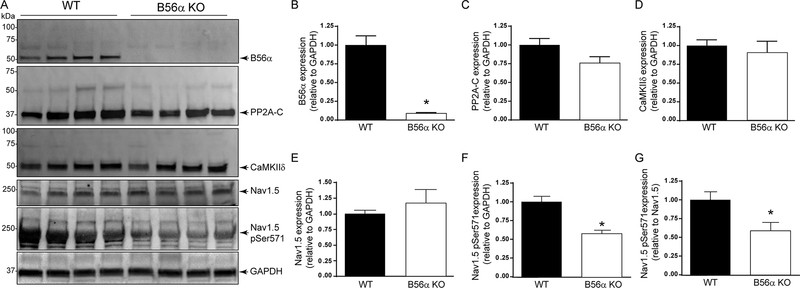
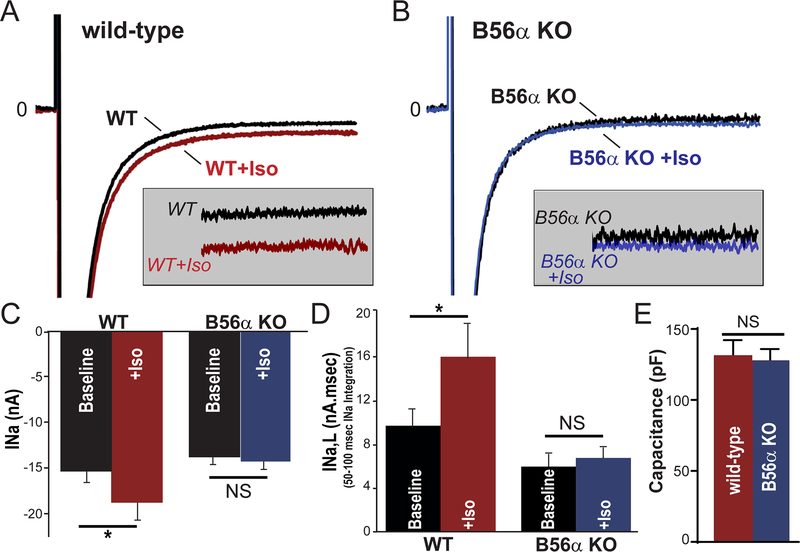
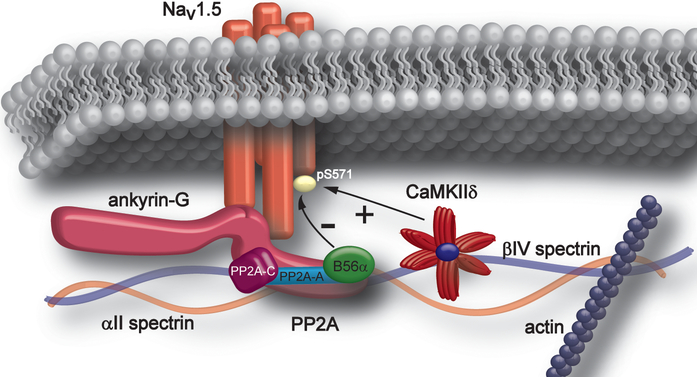
Methods and results: A mouse model lacking a key regulatory subunit (B56α) of the PP (protein phosphatase) 2A holoenzyme displayed aberrant action potentials after adrenergic stimulation. Unbiased computational modeling of B56α KO (knockout) mouse myocyte action potentials revealed an unexpected role of PP2A in INa,L regulation that was confirmed by direct INa,L recordings from B56α KO myocytes. Further, B56α KO myocytes display decreased sensitivity to isoproterenol-induced induction of arrhythmogenic INa,L, and reduced CaMKII-dependent phosphorylation of Nav1.5. At the molecular level, PP2A/B56α complex was found to localize and coimmunoprecipitate with the primary cardiac Nav channel, Nav1.5.
Conclusions: PP2A regulates Nav1.5 activity in mouse cardiomyocytes. This regulation is critical for pathogenic Nav1.5 late current and requires PP2A-B56α. Our study supports B56α as a novel target for the treatment of arrhythmia.
Keywords: ankyrins; arrhythmias, cardiac; calcium-calmodulin-dependent protein kinase type 2; phosphorylation; physiology.
Conflict of interest statement
DISCLOSURES
XHTW is a founding partner of Elex Biotech, a start-up company that developed drug molecules that target ryanodine receptors for the treatment of cardiac arrhythmia disorders. Other authors have no conflicts.
Figures
Comment in
-
Cardiac Sodium Current Under Sympathetic ControlProtein Phosphatase 2A Regulates Cardiac Na+ Channels.Circ Res. 2019 Mar;124(5):674-676. doi: 10.1161/CIRCRESAHA.119.314680. Circ Res. 2019. PMID: 30817259 No abstract available.
-
Response by El Refaey et al to Letter Regarding Article, "Protein Phosphatase 2A Regulates Cardiac Na+ Channels".Circ Res. 2019 Apr 12;124(8):e60-e61. doi: 10.1161/CIRCRESAHA.119.314938. Circ Res. 2019. PMID: 30973807 Free PMC article. No abstract available.
-
Letter by Cristóbal et al Regarding Article, "Protein Phosphatase 2A Regulates Cardiac Na+ Channels".Circ Res. 2019 Apr 12;124(8):e59. doi: 10.1161/CIRCRESAHA.119.314905. Circ Res. 2019. PMID: 30973817 No abstract available.
Similar articles
-
Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function In Vivo.Circulation. 2015 Aug 18;132(7):567-77. doi: 10.1161/CIRCULATIONAHA.114.015218. Epub 2015 Jul 17. Circulation. 2015. PMID: 26187182 Free PMC article.
-
Ca2+/calmodulin-dependent kinase II-dependent regulation of atrial myocyte late Na+ current, Ca2+ cycling, and excitability: a mathematical modeling study.Am J Physiol Heart Circ Physiol. 2017 Dec 1;313(6):H1227-H1239. doi: 10.1152/ajpheart.00185.2017. Epub 2017 Aug 25. Am J Physiol Heart Circ Physiol. 2017. PMID: 28842436 Free PMC article.
-
miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2.Circ Res. 2009 Feb 27;104(4):514-21. doi: 10.1161/CIRCRESAHA.108.181651. Epub 2009 Jan 8. Circ Res. 2009. PMID: 19131648 Free PMC article.
-
Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias.Am J Physiol Heart Circ Physiol. 2013 Aug 15;305(4):H431-45. doi: 10.1152/ajpheart.00306.2013. Epub 2013 Jun 14. Am J Physiol Heart Circ Physiol. 2013. PMID: 23771687 Free PMC article. Review.
-
The arrhythmogenic consequences of increasing late INa in the cardiomyocyte.Cardiovasc Res. 2013 Sep 1;99(4):600-11. doi: 10.1093/cvr/cvt145. Epub 2013 Jun 10. Cardiovasc Res. 2013. PMID: 23752976 Free PMC article. Review.
Cited by
-
Calcium/calmodulin-dependent protein kinase II associates with the K+ channel isoform Kv4.3 in adult rat optic nerve.Front Neuroanat. 2022 Sep 8;16:958986. doi: 10.3389/fnana.2022.958986. eCollection 2022. Front Neuroanat. 2022. PMID: 36172564 Free PMC article.
-
CaMKII in Regulation of Cell Death During Myocardial Reperfusion Injury.Front Mol Biosci. 2021 Jun 1;8:668129. doi: 10.3389/fmolb.2021.668129. eCollection 2021. Front Mol Biosci. 2021. PMID: 34141722 Free PMC article. Review.
-
Abnormal phosphorylation / dephosphorylation and Ca2+ dysfunction in heart failure.Heart Fail Rev. 2024 Jul;29(4):751-768. doi: 10.1007/s10741-024-10395-w. Epub 2024 Mar 18. Heart Fail Rev. 2024. PMID: 38498262 Review.
-
A Novel Gene Signature to Predict Survival Time and Incident Ventricular Arrhythmias in Patients with Dilated Cardiomyopathy.Dis Markers. 2020 Sep 15;2020:8847635. doi: 10.1155/2020/8847635. eCollection 2020. Dis Markers. 2020. PMID: 33014188 Free PMC article.
-
Protein Phosphatase 2A Deficiency in Macrophages Increases Foam Cell Formation and Accelerates Atherosclerotic Lesion Development.Front Cardiovasc Med. 2022 Jan 18;8:745009. doi: 10.3389/fcvm.2021.745009. eCollection 2021. Front Cardiovasc Med. 2022. PMID: 35118139 Free PMC article.
References
-
- Chen-Izu Y, Shaw RM, Pitt GS, Yarov-Yarovoy V, Sack JT, Abriel H, Aldrich RW, Belardinelli L, Cannell MB, Catterall WA, Chazin WJ, Chiamvimonvat N, Deschenes I, Grandi E, Hund TJ, Izu LT, Maier LS, Maltsev VA, Marionneau C, Mohler PJ, Rajamani S, Rasmusson RL, Sobie EA, Clancy CE and Bers DM. Na+ channel function, regulation, structure, trafficking and sequestration. J Physiol. 2015;593:1347–60. - PMC - PubMed
-
- Abriel H Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2009. - PubMed
-
- Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M and Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52. - PubMed
-
- Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, Sag CM, Hund TJ, Mohler PJ, Belardinelli L, Hasenfuss G, Maier LS and Sossalla S. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–22. - PMC - PubMed
-
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ and Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL083422/HL/NHLBI NIH HHS/United States
- R01 HL091947/HL/NHLBI NIH HHS/United States
- R01 HL089598/HL/NHLBI NIH HHS/United States
- R01 HL134824/HL/NHLBI NIH HHS/United States
- R01 HL114383/HL/NHLBI NIH HHS/United States
- R35 HL135754/HL/NHLBI NIH HHS/United States
- T32 HL098039/HL/NHLBI NIH HHS/United States
- R01 HL139348/HL/NHLBI NIH HHS/United States
- F30 HL137331/HL/NHLBI NIH HHS/United States
- R01 HL135096/HL/NHLBI NIH HHS/United States
- R01 HL117641/HL/NHLBI NIH HHS/United States
- R01 HL114893/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
