

Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype
- PMID: 30533528
- PMCID: PMC6244021
- DOI: 10.1212/NXG.0000000000000282
Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype
Abstract
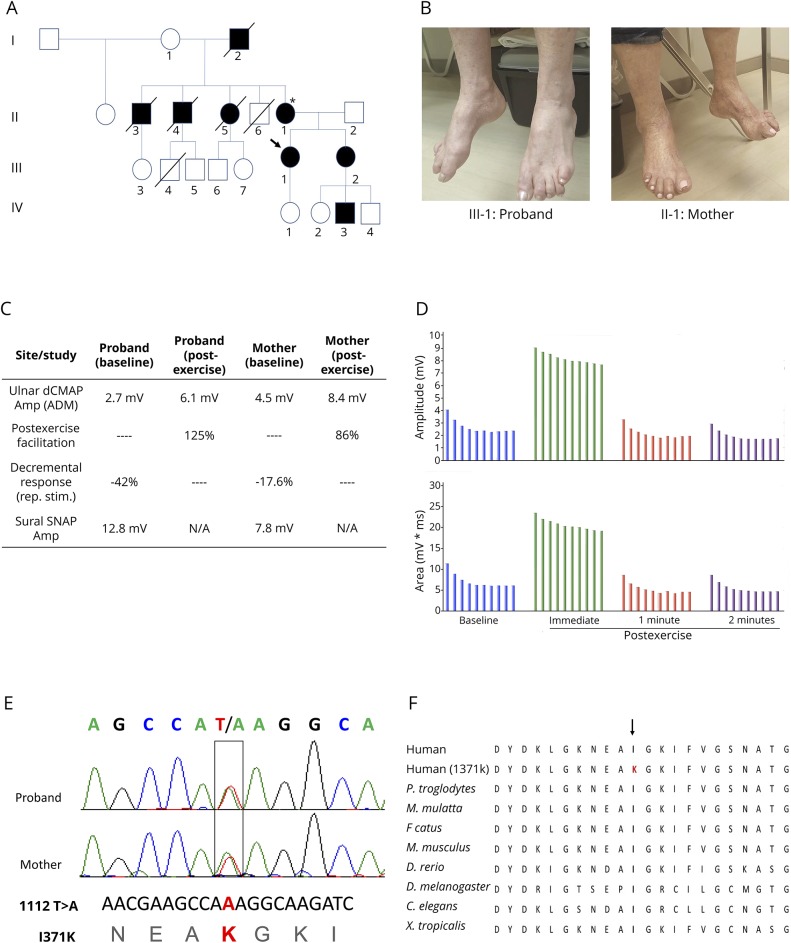
Objective: To report a new SYT2 missense mutation causing distal hereditary motor neuropathy and presynaptic neuromuscular junction (NMJ) transmission dysfunction.
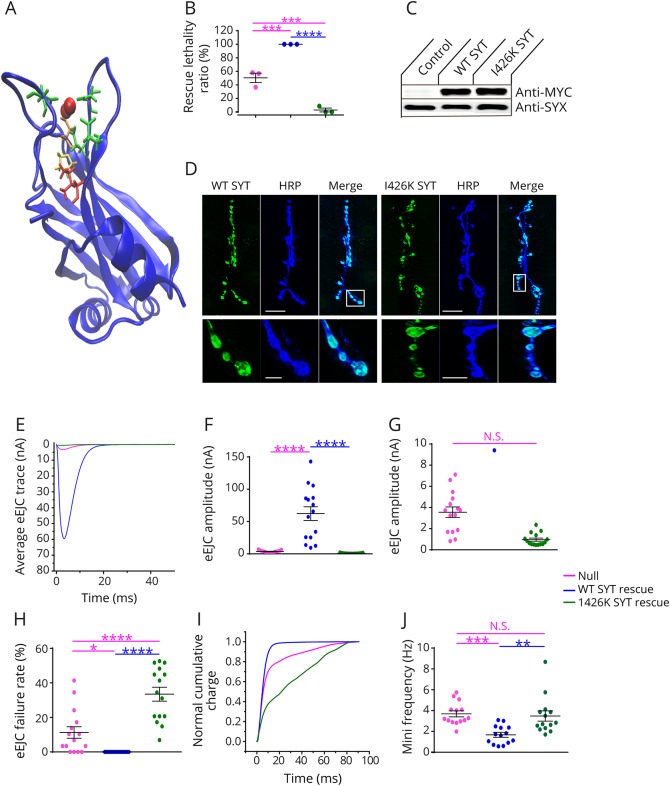
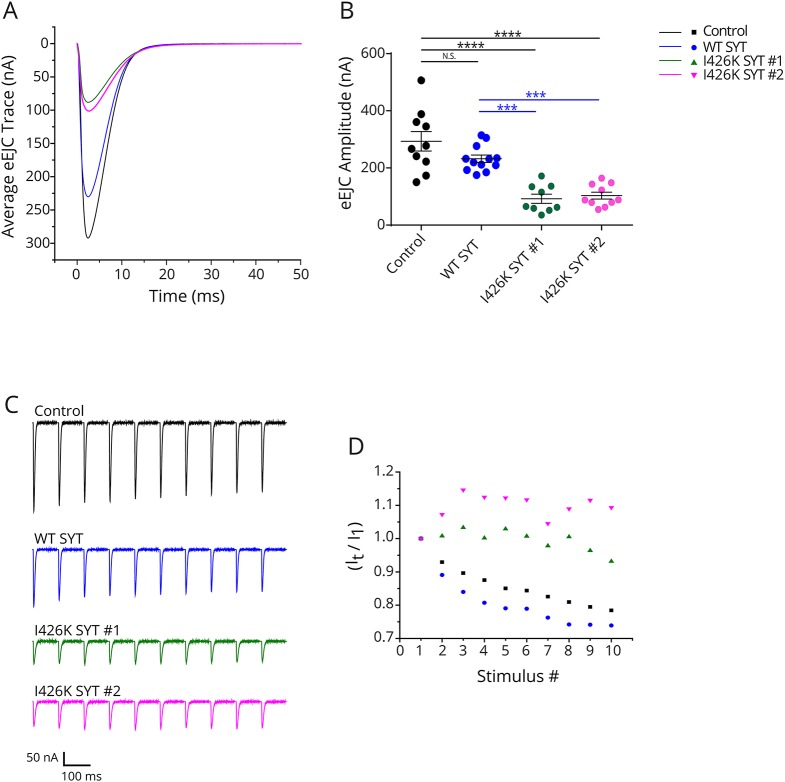
Methods: We report a multigenerational family with a new missense mutation, c. 1112T>A (p. Ile371Lys), in the C2B domain of SYT2, describe the clinical and electrophysiologic phenotype associated with this variant, and validate its pathogenicity in a Drosophila model.
Results: Both proband and her mother present a similar clinical phenotype characterized by a slowly progressive, predominantly motor neuropathy and clear evidence of presynaptic NMJ dysfunction on nerve conduction studies. Validation of this new variant was accomplished by characterization of the mutation homologous to the human c. 1112T>A variant in Drosophila, confirming its dominant-negative effect on neurotransmitter release.
Conclusions: This report provides further confirmation of the role of SYT2 in human disease and corroborates the resultant unique clinical phenotype consistent with heriditary distal motor neuropathy. SYT2-related motor neuropathy is a rare disease but should be suspected in patients presenting with a combination of presynaptic NMJ dysfunction (resembling Lambert-Eaton myasthenic syndrome) and a predominantly motor neuropathy, especially in the context of a positive family history.
Figures
Similar articles
-
Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome.Am J Med Genet A. 2020 Oct;182(10):2272-2283. doi: 10.1002/ajmg.a.61765. Epub 2020 Aug 10. Am J Med Genet A. 2020. PMID: 32776697 Free PMC article.
-
Identification of a Novel de Novo Variant in the SYT2 Gene Causing a Rare Type of Distal Hereditary Motor Neuropathy.Genes (Basel). 2020 Oct 22;11(11):1238. doi: 10.3390/genes11111238. Genes (Basel). 2020. PMID: 33105646 Free PMC article.
-
A new de novo SYT2 mutation presenting as distal weakness. Neuropathy or neuromuscular junction dysfunction?J Peripher Nerv Syst. 2021 Mar;26(1):113-117. doi: 10.1111/jns.12425. Epub 2020 Dec 22. J Peripher Nerv Syst. 2021. PMID: 33320396
-
How to Spot Congenital Myasthenic Syndromes Resembling the Lambert-Eaton Myasthenic Syndrome? A Brief Review of Clinical, Electrophysiological, and Genetics Features.Neuromolecular Med. 2018 Jun;20(2):205-214. doi: 10.1007/s12017-018-8490-1. Epub 2018 Apr 25. Neuromolecular Med. 2018. PMID: 29696584 Review.
-
Lambert-Eaton Myasthenic Syndrome.Neurol Clin. 2018 May;36(2):379-394. doi: 10.1016/j.ncl.2018.01.008. Neurol Clin. 2018. PMID: 29655456 Free PMC article. Review.
Cited by
-
Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome.Am J Med Genet A. 2020 Oct;182(10):2272-2283. doi: 10.1002/ajmg.a.61765. Epub 2020 Aug 10. Am J Med Genet A. 2020. PMID: 32776697 Free PMC article.
-
Early onset hereditary neuronopathies: an update on non-5q motor neuron diseases.Brain. 2023 Mar 1;146(3):806-822. doi: 10.1093/brain/awac452. Brain. 2023. PMID: 36445400 Free PMC article. Review.
-
The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms.Front Neurol. 2019 Mar 19;10:257. doi: 10.3389/fneur.2019.00257. eCollection 2019. Front Neurol. 2019. PMID: 30941097 Free PMC article. Review.
-
New recessive mutations in SYT2 causing severe presynaptic congenital myasthenic syndromes.Neurol Genet. 2020 Dec 3;6(6):e534. doi: 10.1212/NXG.0000000000000534. eCollection 2020 Dec. Neurol Genet. 2020. PMID: 33659639 Free PMC article.
-
Similarity and Diversity of Presynaptic Molecules at Neuromuscular Junctions and Central Synapses.Biomolecules. 2022 Jan 21;12(2):179. doi: 10.3390/biom12020179. Biomolecules. 2022. PMID: 35204679 Free PMC article. Review.
References
-
- Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 2012;83:6–14. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials