

Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations
- PMID: 30533525
- PMCID: PMC6244025
- DOI: 10.1212/NXG.0000000000000279
Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations
Erratum in
-
Erratum: Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations.Neurol Genet. 2018 Dec 3;4(6):e300. doi: 10.1212/NXG.0000000000000300. eCollection 2018 Dec. Neurol Genet. 2018. PMID: 30588500 Free PMC article.
Abstract
Objective: To clinically, genetically, and radiologically characterize a large cohort of SPG7 patients.
Methods: We used data from next-generation sequencing panels for ataxias and hereditary spastic paraplegia to identify a characteristic phenotype that helped direct genetic testing for variations in SPG7. We analyzed MRI. We reviewed all published SPG7 mutations for correlations.
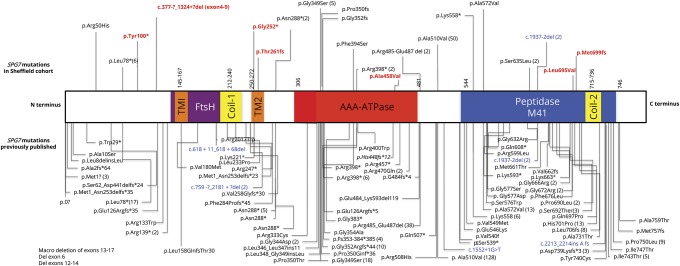
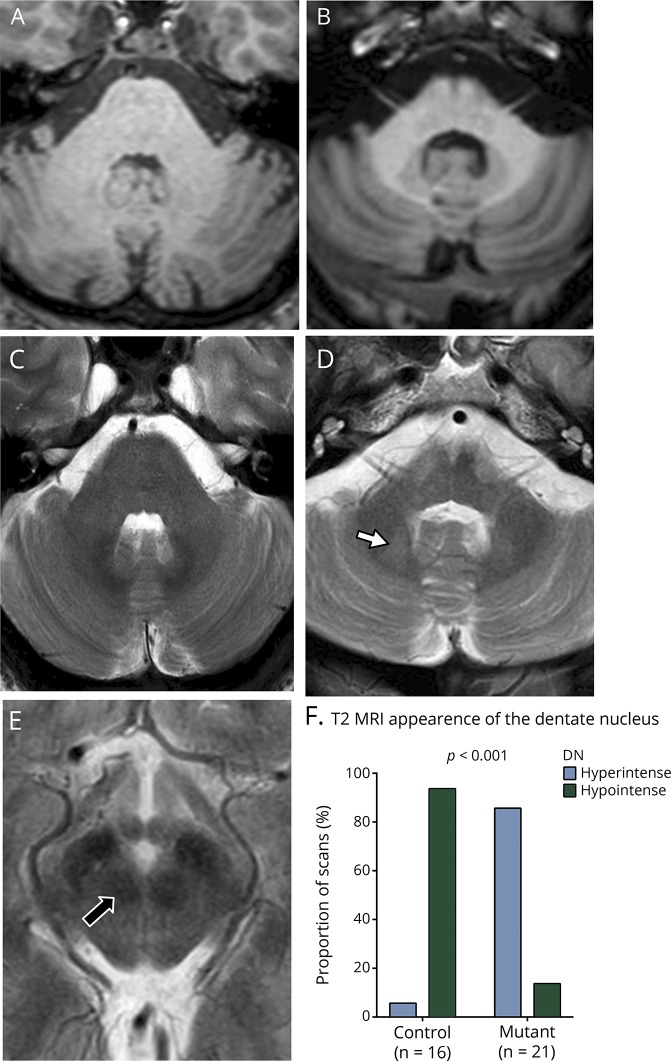
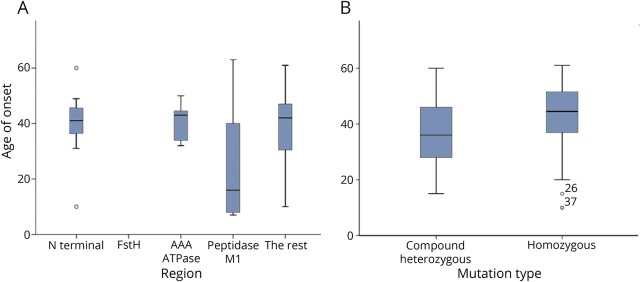
Results: We identified 42 cases with biallelic SPG7 mutations, including 7 novel mutations, including a large multi-exon deletion, representing one of the largest cohorts so far described. We identified a characteristic phenotype comprising cerebellar ataxia with prominent cerebellar dysarthria, mild lower limb spasticity, and a waddling gait, predominantly from a cohort of idiopathic ataxia. We report a rare brain MRI finding of dentate nucleus hyperintensity on T2 sequences with SPG7 mutations. We confirm that the c.1529C>T allele is frequently present in patients with long-standing British ancestry. Based on the findings of the present study and existing literature, we confirm that patients with homozygous mutations involving the M41 peptidase domain of SPG7 have a younger age at onset compared to individuals with mutations elsewhere in the gene (14 years difference, p < 0.034), whereas c.1529C>T compound heterozygous mutations are associated with a younger age at onset compared to homozygous cases (5.4 years difference, p < 0.022).
Conclusions: Mutant SPG7 is common in sporadic ataxia. In patients with British ancestry, c.1529C>T allele represents the most frequent mutation. SPG7 mutations can be clinically predicted by the characteristic hybrid spastic-ataxic phenotype described above, along with T2 hyperintensity of the dentate nucleus on MRI.
Figures
Similar articles
-
Prevalence and phenotype of the c.1529C>T SPG7 variant in adult-onset cerebellar ataxia in Italy.Eur J Neurol. 2019 Jan;26(1):80-86. doi: 10.1111/ene.13768. Epub 2018 Sep 3. Eur J Neurol. 2019. PMID: 30098094
-
Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy.Brain. 2012 Oct;135(Pt 10):2980-93. doi: 10.1093/brain/aws240. Brain. 2012. PMID: 23065789 Free PMC article.
-
Clinical and genetic characteristics of 21 Spanish patients with biallelic pathogenic SPG7 mutations.J Neurol Sci. 2021 Oct 15;429:118062. doi: 10.1016/j.jns.2021.118062. Epub 2021 Aug 30. J Neurol Sci. 2021. PMID: 34500365
-
Genotype-phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients.J Neurol. 2021 Jun;268(6):2065-2082. doi: 10.1007/s00415-019-09633-1. Epub 2019 Nov 19. J Neurol. 2021. PMID: 31745725 Review.
-
Spastic paraplegia type 46: novel and recurrent GBA2 gene variants in a compound heterozygous Italian patient with spastic ataxia phenotype.Neurol Sci. 2021 Nov;42(11):4741-4745. doi: 10.1007/s10072-021-05463-0. Epub 2021 Jul 12. Neurol Sci. 2021. PMID: 34251556 Review.
Cited by
-
Clinico-Genetic, Imaging and Molecular Delineation of COQ8A-Ataxia: A Multicenter Study of 59 Patients.Ann Neurol. 2020 Aug;88(2):251-263. doi: 10.1002/ana.25751. Epub 2020 Jun 10. Ann Neurol. 2020. PMID: 32337771 Free PMC article.
-
SPG7 mutations in amyotrophic lateral sclerosis: a genetic link to hereditary spastic paraplegia.J Neurol. 2020 Sep;267(9):2732-2743. doi: 10.1007/s00415-020-09861-w. Epub 2020 May 23. J Neurol. 2020. PMID: 32447552 Free PMC article.
-
Unravelling the etiology of sporadic late-onset cerebellar ataxia in a cohort of 205 patients: a prospective study.J Neurol. 2022 Dec;269(12):6354-6365. doi: 10.1007/s00415-022-11253-1. Epub 2022 Jul 23. J Neurol. 2022. PMID: 35869996
-
Erratum: Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations.Neurol Genet. 2018 Dec 3;4(6):e300. doi: 10.1212/NXG.0000000000000300. eCollection 2018 Dec. Neurol Genet. 2018. PMID: 30588500 Free PMC article.
-
Episodic ataxia and severe infantile phenotype in spinocerebellar ataxia type 14: expansion of the phenotype and novel mutations.J Neurol. 2022 Mar;269(3):1476-1484. doi: 10.1007/s00415-021-10712-5. Epub 2021 Jul 22. J Neurol. 2022. PMID: 34292398 Free PMC article.
References
-
- Refsum S, Skre H. Neurological approaches to the inherited ataxias. Adv Neurol 1978;21:1–13. - PubMed
-
- Polo JM, Calleja J, Combarros O, Berciano J. Hereditary ataxias and paraplegias in Cantabria, Spain. An epidemiological and clinical study. Brain 1991;114(pt 2):855–866. - PubMed
-
- McDermott CJ, Shaw PJ. Chapter 17 Hereditary spastic paraparesis. Handb Clin Neurol 2007;82:327–352. - PubMed
-
- Matilla-Duenas A. The ever expanding spinocerebellar ataxias. Editorial. Cerebellum 2012;11:821–827. - PubMed
-
- Campanella G, Filla A, De Michele G. Classifications of hereditary ataxias: a critical overview. Acta Neurol (Napoli) 1992;14:408–419. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Medical