

Origin and evolution of pathogenic coronaviruses
- PMID: 30531947
- PMCID: PMC7097006
- DOI: 10.1038/s41579-018-0118-9
Origin and evolution of pathogenic coronaviruses
Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) are two highly transmissible and pathogenic viruses that emerged in humans at the beginning of the 21st century. Both viruses likely originated in bats, and genetically diverse coronaviruses that are related to SARS-CoV and MERS-CoV were discovered in bats worldwide. In this Review, we summarize the current knowledge on the origin and evolution of these two pathogenic coronaviruses and discuss their receptor usage; we also highlight the diversity and potential of spillover of bat-borne coronaviruses, as evidenced by the recent spillover of swine acute diarrhoea syndrome coronavirus (SADS-CoV) to pigs.
Conflict of interest statement
The authors declare no competing interests.
Figures
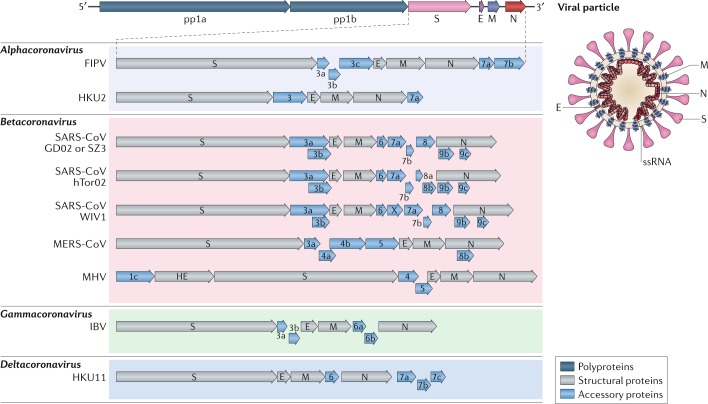

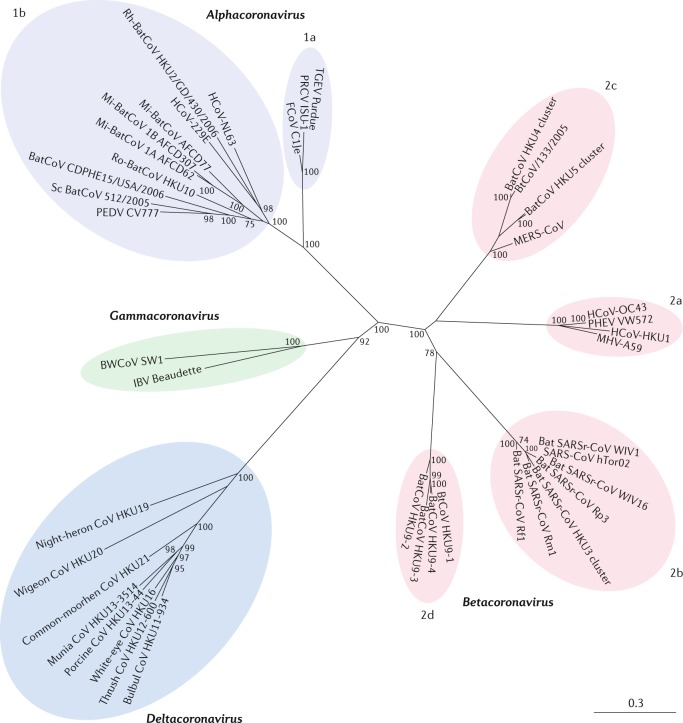
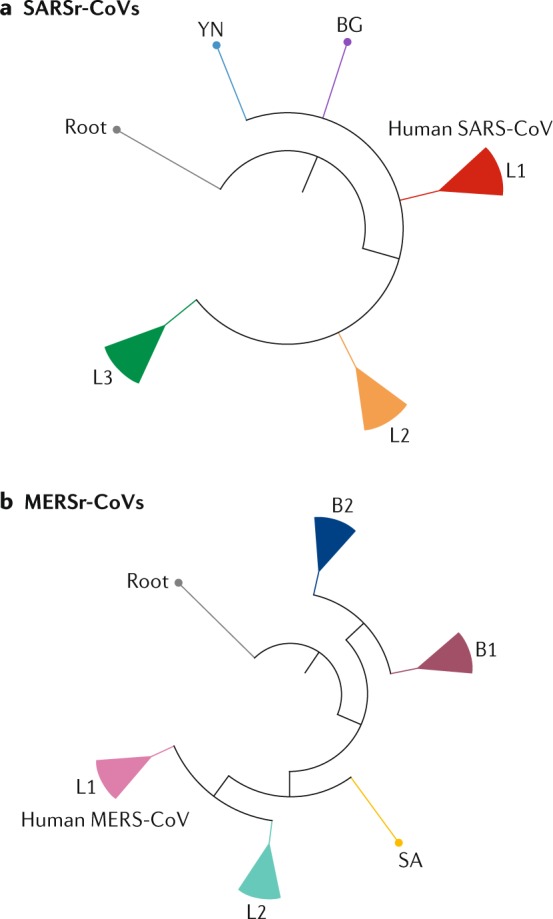

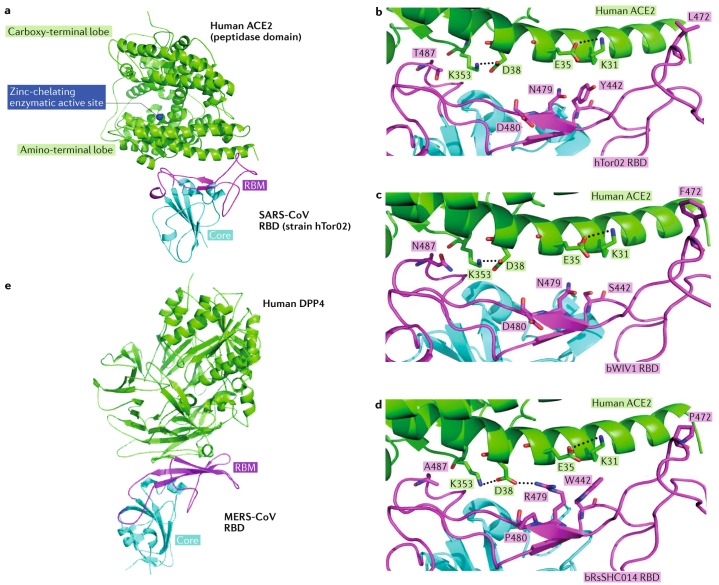
Comment in
-
Commentary: Origin and evolution of pathogenic coronaviruses.Front Immunol. 2020 Apr 21;11:811. doi: 10.3389/fimmu.2020.00811. eCollection 2020. Front Immunol. 2020. PMID: 32373134 Free PMC article. No abstract available.
Similar articles
-
Broad Cross-Species Infection of Cultured Cells by Bat HKU2-Related Swine Acute Diarrhea Syndrome Coronavirus and Identification of Its Replication in Murine Dendritic Cells In Vivo Highlight Its Potential for Diverse Interspecies Transmission.J Virol. 2019 Nov 26;93(24):e01448-19. doi: 10.1128/JVI.01448-19. Print 2019 Dec 15. J Virol. 2019. PMID: 31554686 Free PMC article.
-
SARS-Coronavirus ancestor's foot-prints in South-East Asian bat colonies and the refuge theory.Infect Genet Evol. 2011 Oct;11(7):1690-702. doi: 10.1016/j.meegid.2011.06.021. Epub 2011 Jul 8. Infect Genet Evol. 2011. PMID: 21763784 Free PMC article.
-
Comprehensive comparative genomic and microsatellite analysis of SARS, MERS, BAT-SARS, and COVID-19 coronaviruses.J Med Virol. 2021 Jul;93(7):4382-4391. doi: 10.1002/jmv.26974. Epub 2021 Apr 8. J Med Virol. 2021. PMID: 33782990 Free PMC article.
-
Interspecies Jumping of Bat Coronaviruses.Viruses. 2021 Oct 29;13(11):2188. doi: 10.3390/v13112188. Viruses. 2021. PMID: 34834994 Free PMC article. Review.
-
From SARS to MERS, Thrusting Coronaviruses into the Spotlight.Viruses. 2019 Jan 14;11(1):59. doi: 10.3390/v11010059. Viruses. 2019. PMID: 30646565 Free PMC article. Review.
Cited by
-
Design of customized coronavirus receptors.Nature. 2024 Oct 30. doi: 10.1038/s41586-024-08121-5. Online ahead of print. Nature. 2024. PMID: 39478224
-
Hemophagocytosis of the Hilar Pulmonary Lymph Nodes Is a More Sensitive Indicator of the Severity of COVID-19 Disease than Bone Marrow Hemophagocytosis.Diseases. 2024 Oct 3;12(10):241. doi: 10.3390/diseases12100241. Diseases. 2024. PMID: 39452484 Free PMC article.
-
Decoding the intricacies: a comprehensive analysis of microRNAs in the pathogenesis, diagnosis, prognosis and therapeutic strategies for COVID-19.Front Med (Lausanne). 2024 Oct 7;11:1430974. doi: 10.3389/fmed.2024.1430974. eCollection 2024. Front Med (Lausanne). 2024. PMID: 39434774 Free PMC article. Review.
-
COVID-19 and persistent symptoms: implications for polycystic ovary syndrome and its management.Front Endocrinol (Lausanne). 2024 Oct 4;15:1434331. doi: 10.3389/fendo.2024.1434331. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 39429741 Free PMC article. Review.
-
Systematic review and meta analysis of cross immunity and the smokers paradox in COVID19.Sci Rep. 2024 Oct 17;14(1):24344. doi: 10.1038/s41598-024-75632-6. Sci Rep. 2024. PMID: 39420134 Free PMC article.
References
-
- Masters, P. S. & Perlman, S. in Fields Virology Vol. 2 (eds Knipe, D. M. & Howley, P. M.) 825–858 (Lippincott Williams & Wilkins, 2013).
-
- Drosten C, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. - PubMed
-
- Ksiazek TG, et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
