

Cadmium opens GluK2 kainate receptors with cysteine substitutions at the M3 helix bundle crossing
- PMID: 30498132
- PMCID: PMC6445585
- DOI: 10.1085/jgp.201812234
Cadmium opens GluK2 kainate receptors with cysteine substitutions at the M3 helix bundle crossing
Abstract
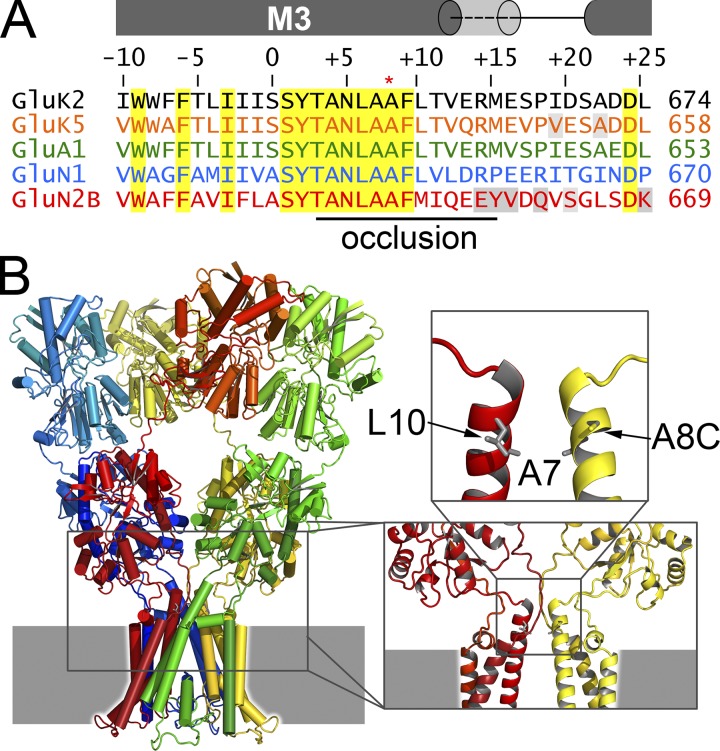
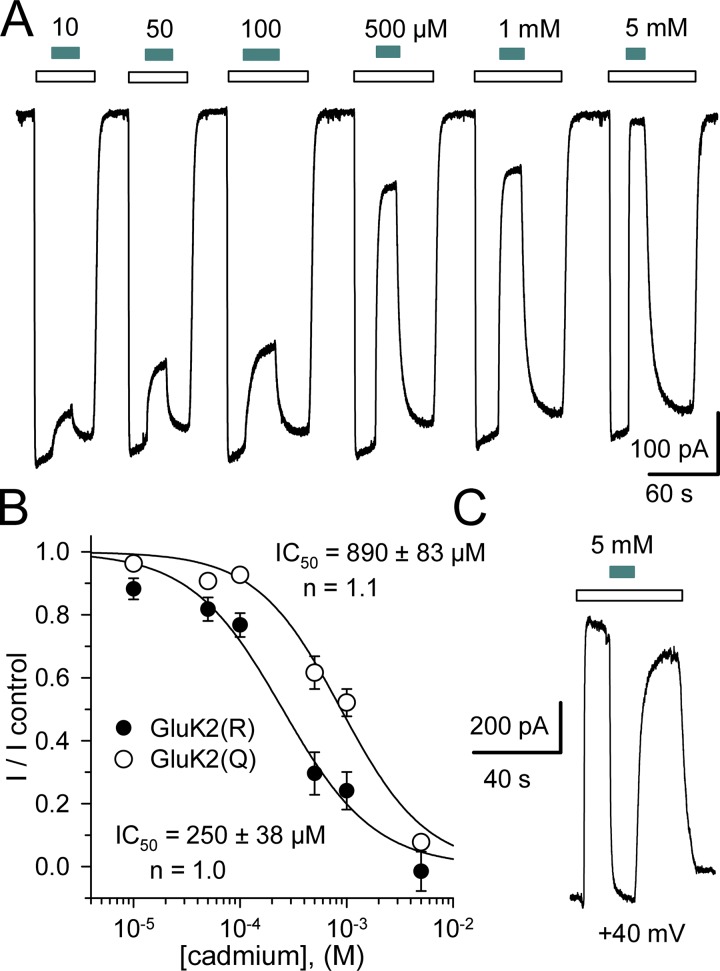
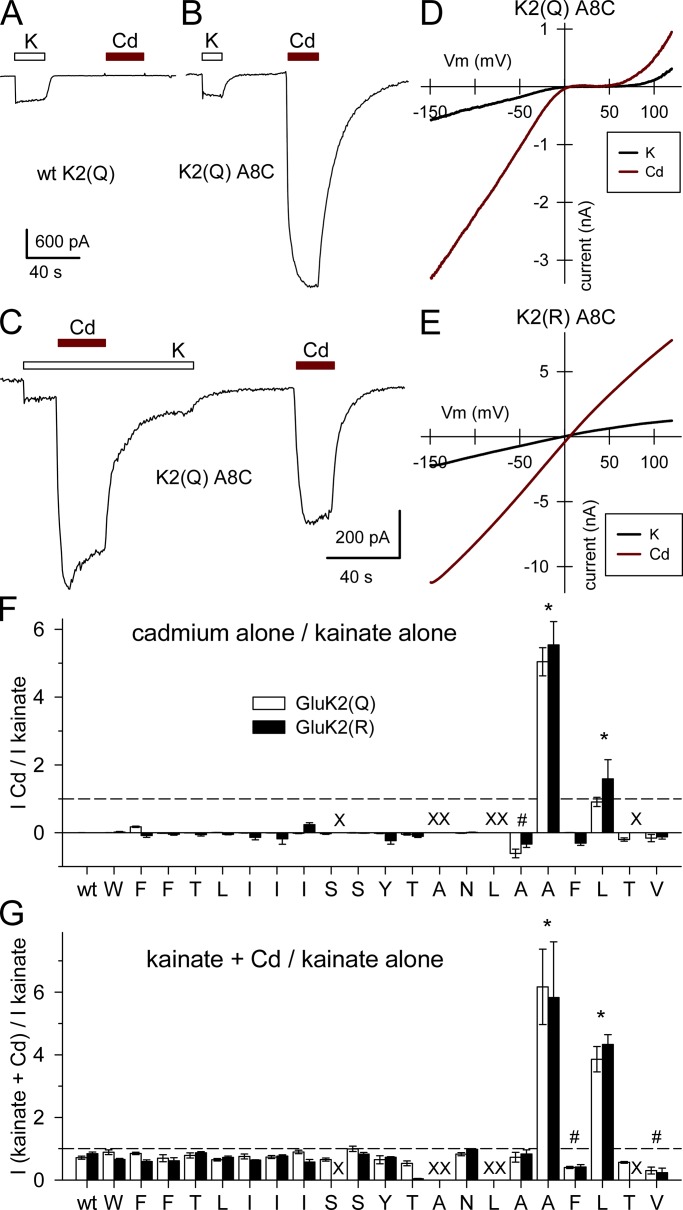
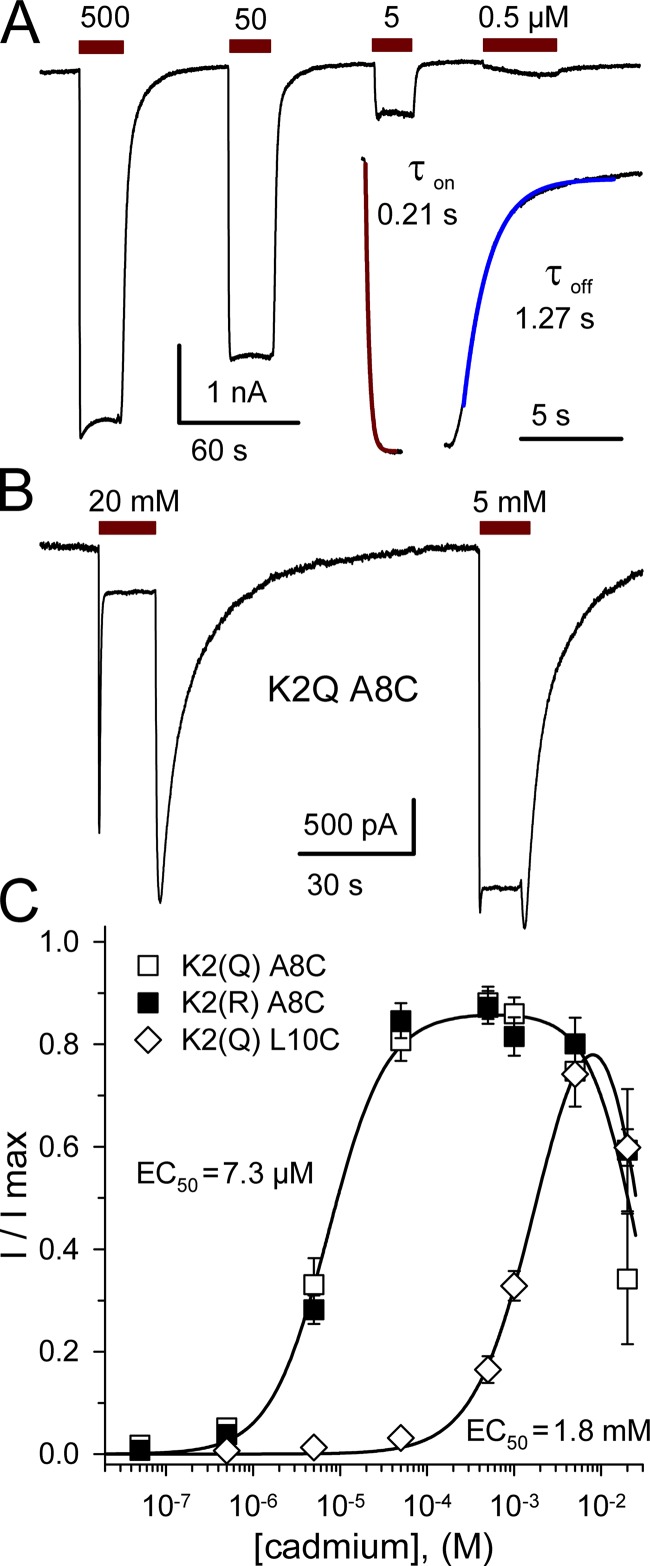
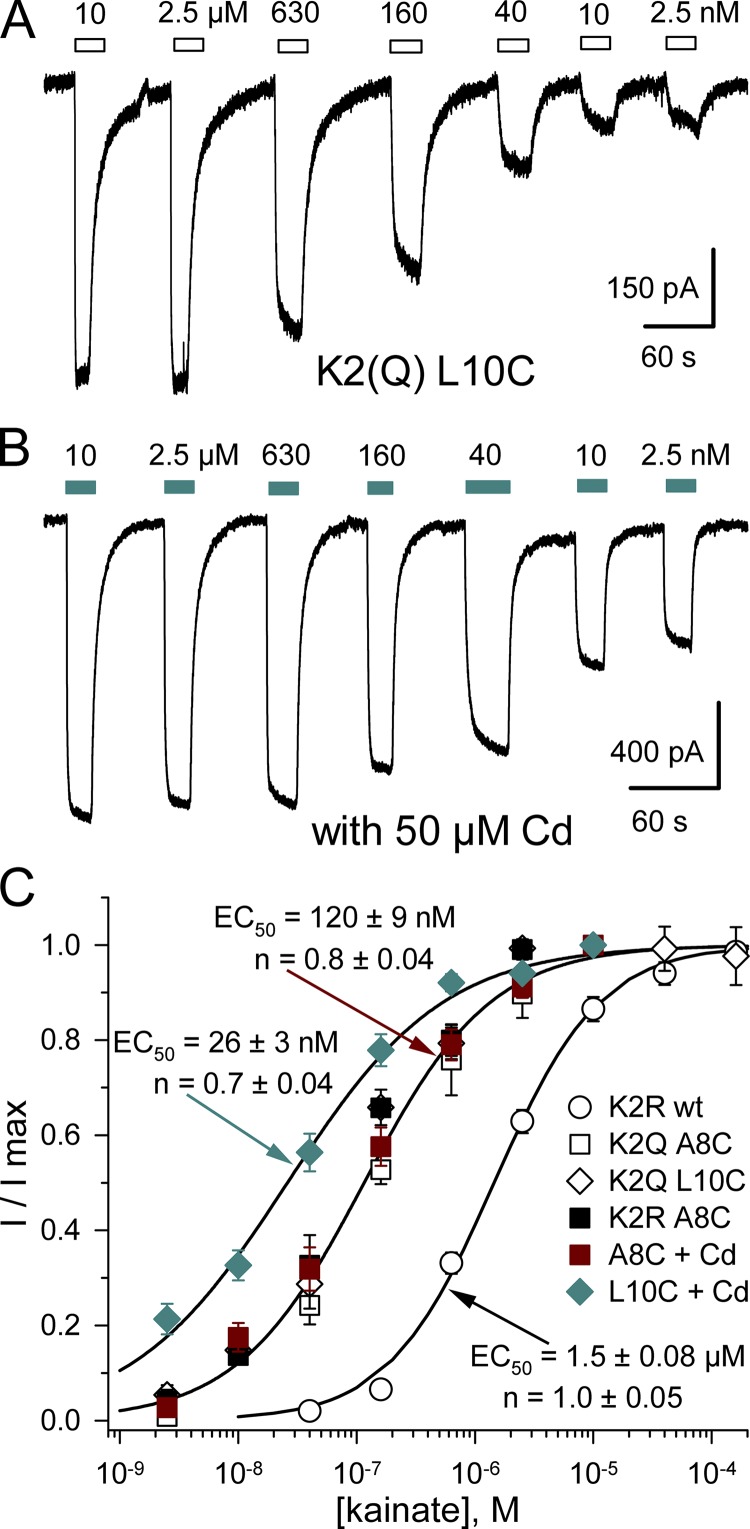
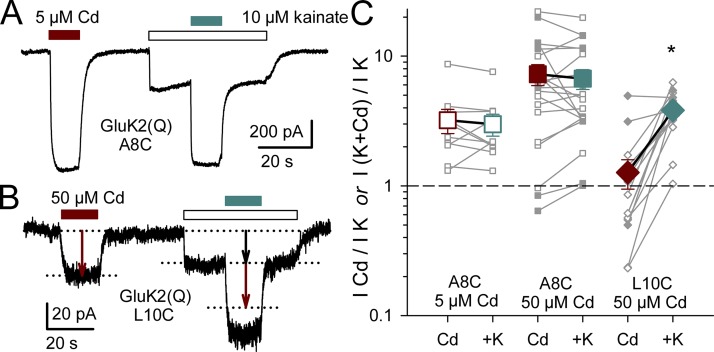
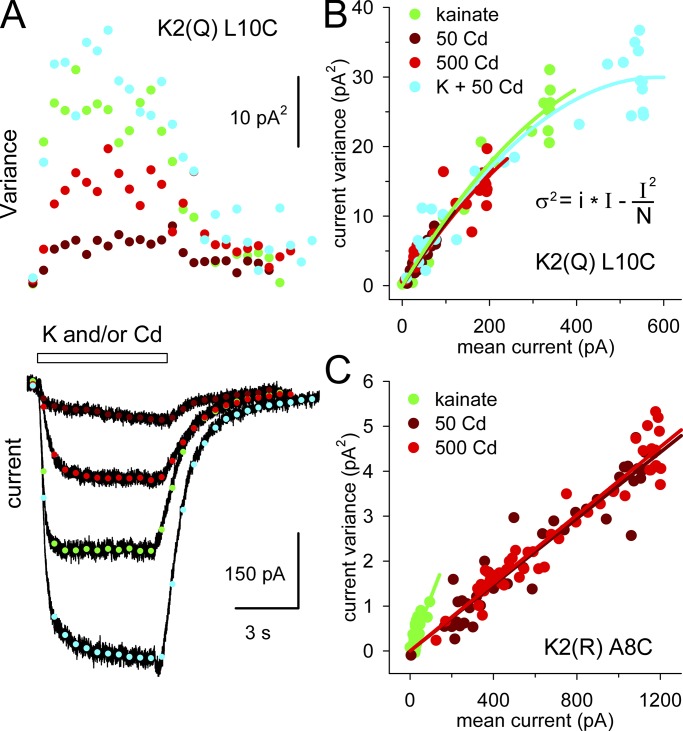
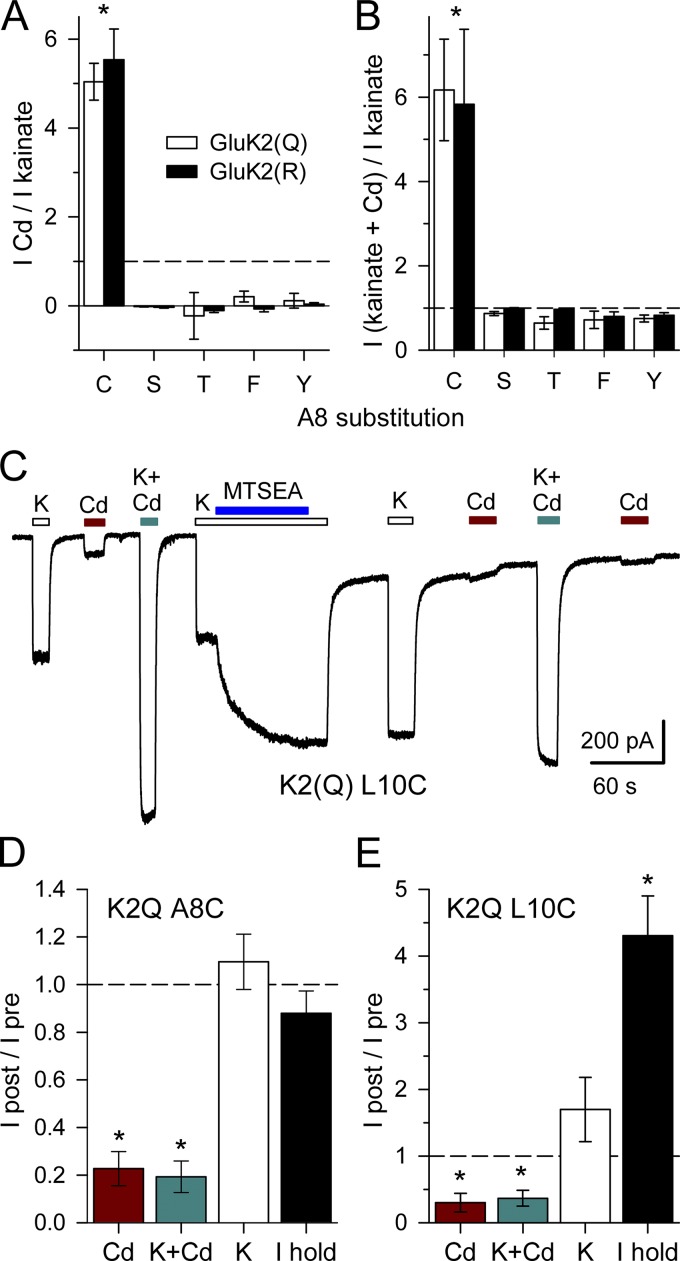
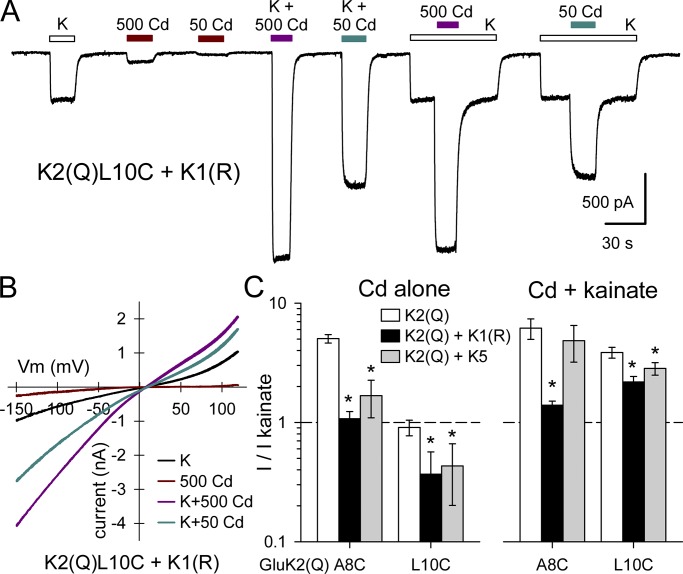
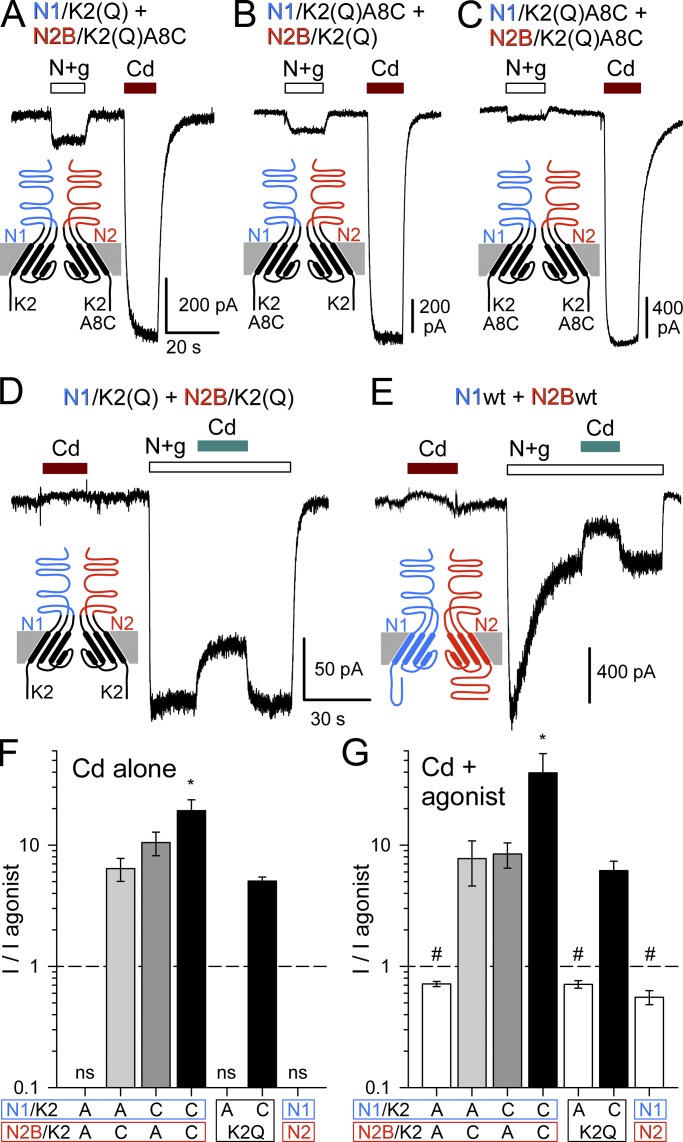
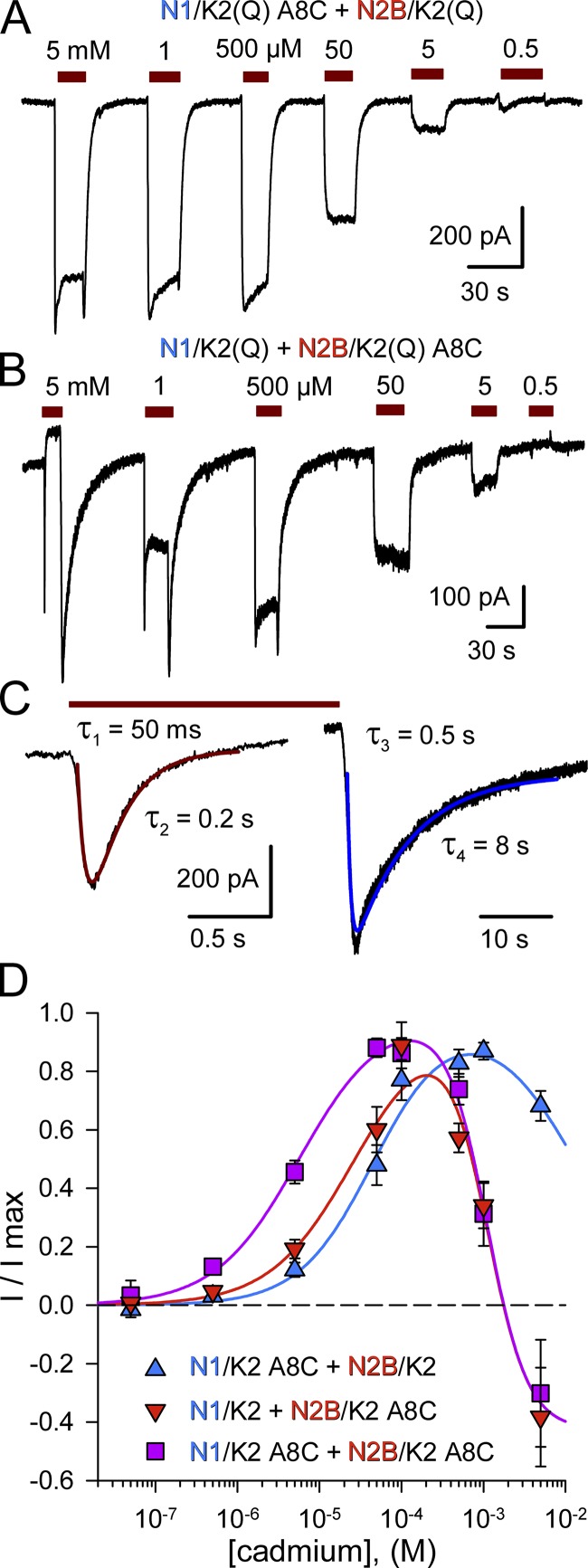
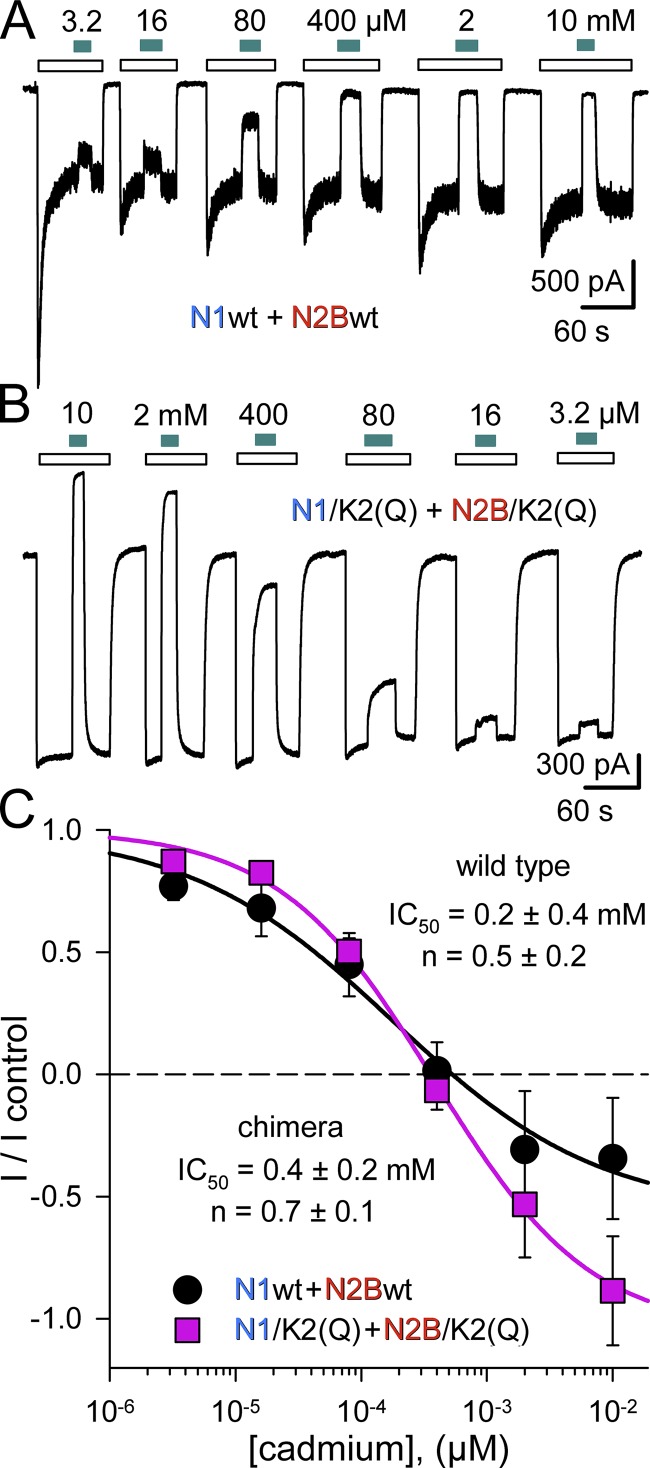
Kainate receptors are ligand-gated ion channels that have two major roles in the central nervous system: they mediate a postsynaptic component of excitatory neurotransmission at some glutamatergic synapses and modulate transmitter release at both excitatory and inhibitory synapses. Accumulating evidence implicates kainate receptors in a variety of neuropathologies, including epilepsy, psychiatric disorders, developmental delay, and cognitive impairment. Here, to gain a deeper understanding of the conformational changes associated with agonist binding and channel opening, we generate a series of Cys substitutions in the GluK2 kainate receptor subunit, focusing on the M3 helices that line the ion pore and form the bundle-crossing gate at the extracellular mouth of the channel. Exposure to 50 µM Cd produces direct activation of homomeric mutant channels bearing Cys substitutions in (A657C), or adjacent to (L659C), the conserved SYTANLAAF motif. Activation by Cd is occluded by modification with 2-aminoethyl MTS (MTSEA), indicating that Cd binds directly and specifically to the substituted cysteines. Cd potency for the A657C mutation (EC50 = 10 µM) suggests that binding involves at least two coordinating residues, whereas weaker Cd potency for L659C (EC50 = 2 mM) implies that activation does not require tight coordination by multiple side chains for this substitution. Experiments with heteromeric and chimeric channels indicate that activation by Cd requires Cys substitution at only two of the four subunits within a tetrameric receptor and that activation is similar for substitution within subunits in either the A/C or B/D conformations. We develop simple kinetic models for the A657C substitution that reproduce several features of Cd activation as well as the low-affinity inhibition observed at higher Cd concentrations (5-20 mM). Together, these results demonstrate rapid and reversible channel activation, independent of agonist site occupancy, upon Cd binding to Cys side chains at two specific locations along the GluK2 inner helix.
© 2019 Wilding and Huettner.
Figures
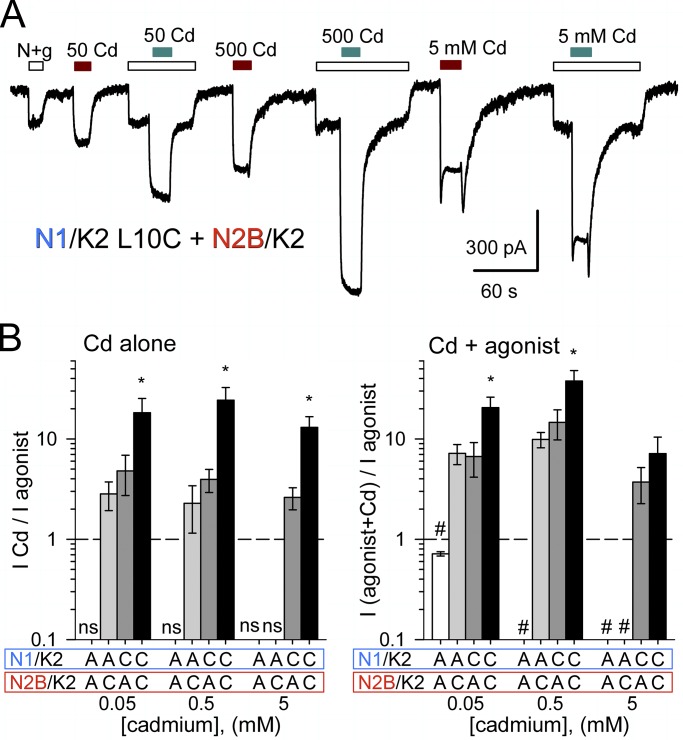
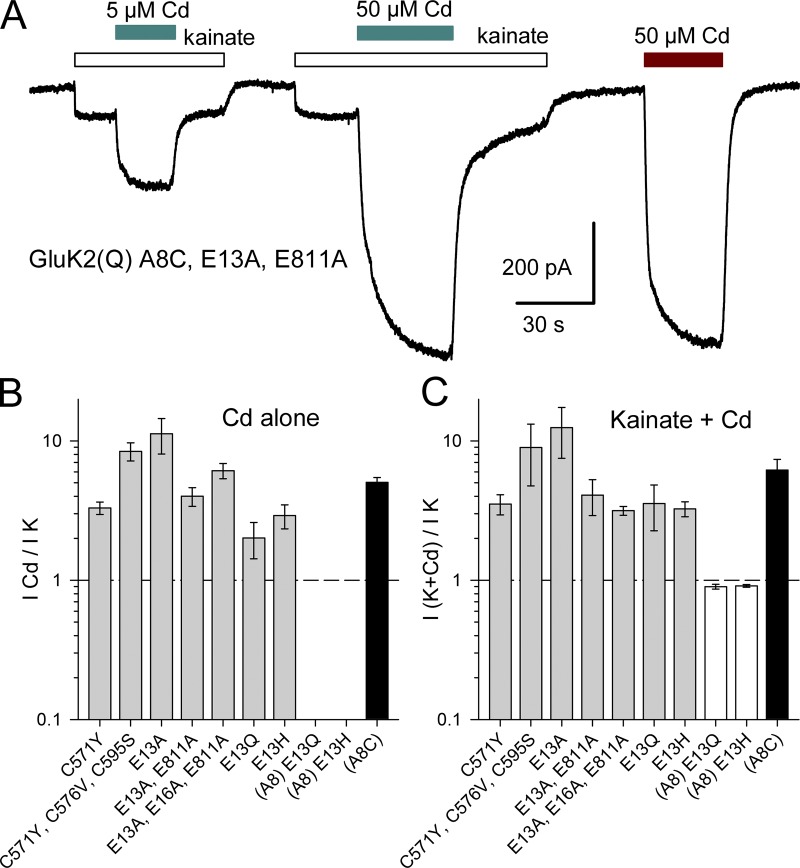
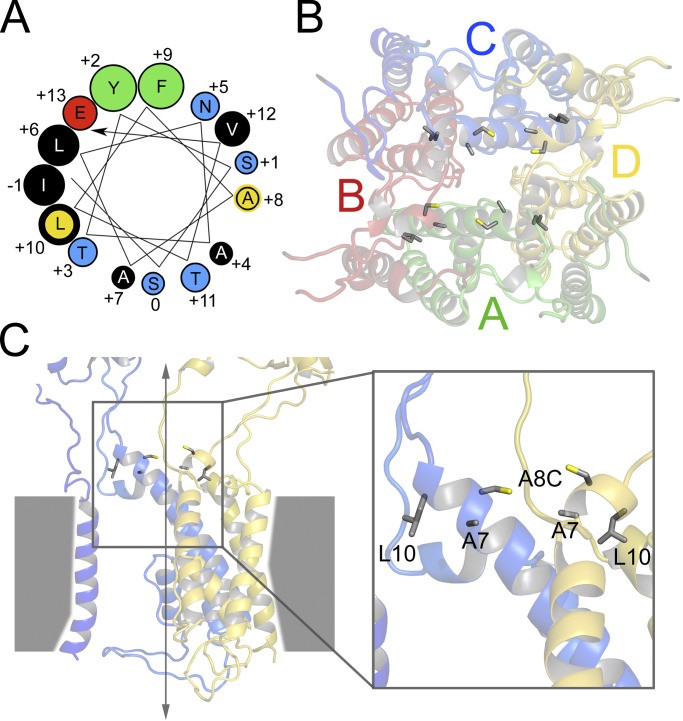
Comment in
-
Prying open a glutamate receptor gate.J Gen Physiol. 2019 Apr 1;151(4):396-399. doi: 10.1085/jgp.201812312. Epub 2019 Feb 28. J Gen Physiol. 2019. PMID: 30819719 Free PMC article.
Similar articles
-
Cadmium activates AMPA and NMDA receptors with M3 helix cysteine substitutions.J Gen Physiol. 2020 Jul 6;152(7):e201912537. doi: 10.1085/jgp.201912537. J Gen Physiol. 2020. PMID: 32342094 Free PMC article.
-
Q/R site interactions with the M3 helix in GluK2 kainate receptor channels revealed by thermodynamic mutant cycles.J Gen Physiol. 2013 Sep;142(3):225-39. doi: 10.1085/jgp.201311000. Epub 2013 Aug 12. J Gen Physiol. 2013. PMID: 23940260 Free PMC article.
-
Fatty acid modulation and polyamine block of GluK2 kainate receptors analyzed by scanning mutagenesis.J Gen Physiol. 2010 Sep;136(3):339-52. doi: 10.1085/jgp.201010442. J Gen Physiol. 2010. PMID: 20805577 Free PMC article.
-
Genetic manipulation of key determinants of ion flow in glutamate receptor channels in the mouse.Brain Res. 2001 Jul 13;907(1-2):233-43. doi: 10.1016/s0006-8993(01)02445-3. Brain Res. 2001. PMID: 11430906 Review.
-
Emerging issues of connexin channels: biophysics fills the gap.Q Rev Biophys. 2001 Aug;34(3):325-472. doi: 10.1017/s0033583501003705. Q Rev Biophys. 2001. PMID: 11838236 Review.
Cited by
-
Correlation between GRIK2 rs6922753, rs2227283 polymorphism and aggressive behaviors with Bipolar Mania in the Chinese Han population.Brain Behav. 2019 Nov;9(11):e01449. doi: 10.1002/brb3.1449. Epub 2019 Oct 20. Brain Behav. 2019. PMID: 31631587 Free PMC article.
-
Prying open a glutamate receptor gate.J Gen Physiol. 2019 Apr 1;151(4):396-399. doi: 10.1085/jgp.201812312. Epub 2019 Feb 28. J Gen Physiol. 2019. PMID: 30819719 Free PMC article.
-
Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels.Pharmacol Rev. 2021 Oct;73(4):298-487. doi: 10.1124/pharmrev.120.000131. Pharmacol Rev. 2021. PMID: 34753794 Free PMC article. Review.
-
Cadmium activates AMPA and NMDA receptors with M3 helix cysteine substitutions.J Gen Physiol. 2020 Jul 6;152(7):e201912537. doi: 10.1085/jgp.201912537. J Gen Physiol. 2020. PMID: 32342094 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
