

Roles of the inflammasome in the gut‑liver axis (Review)
- PMID: 30483776
- PMCID: PMC6297761
- DOI: 10.3892/mmr.2018.9679
Roles of the inflammasome in the gut‑liver axis (Review)
Abstract
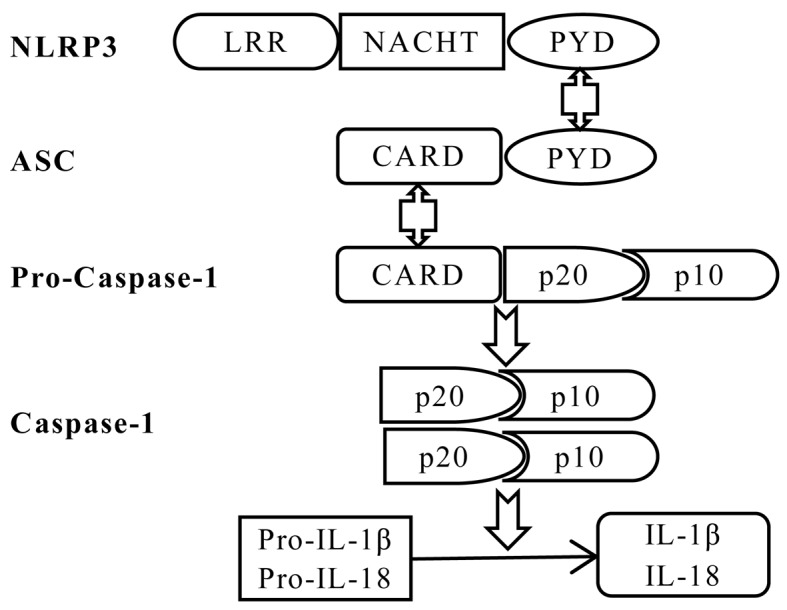
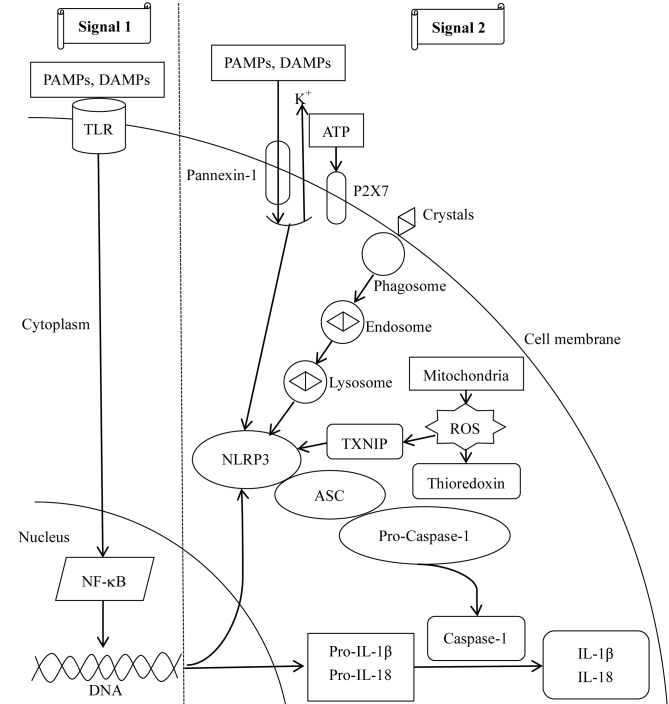
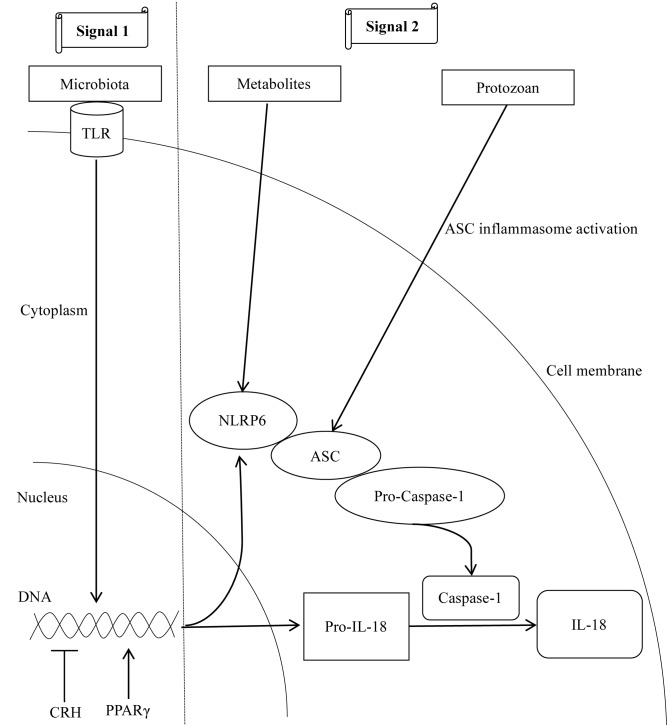
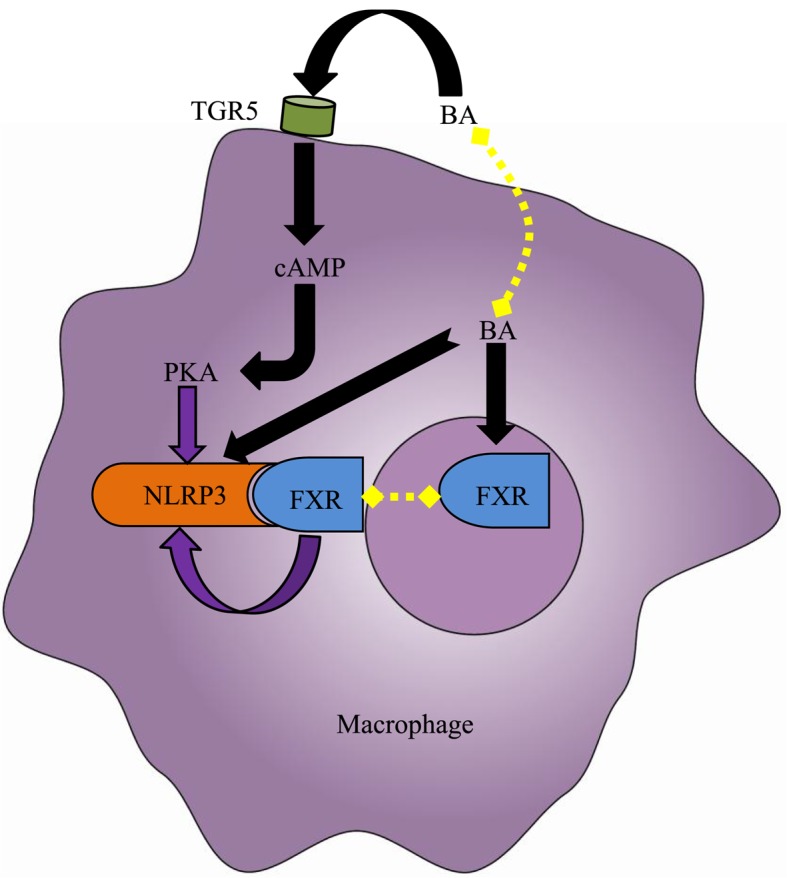
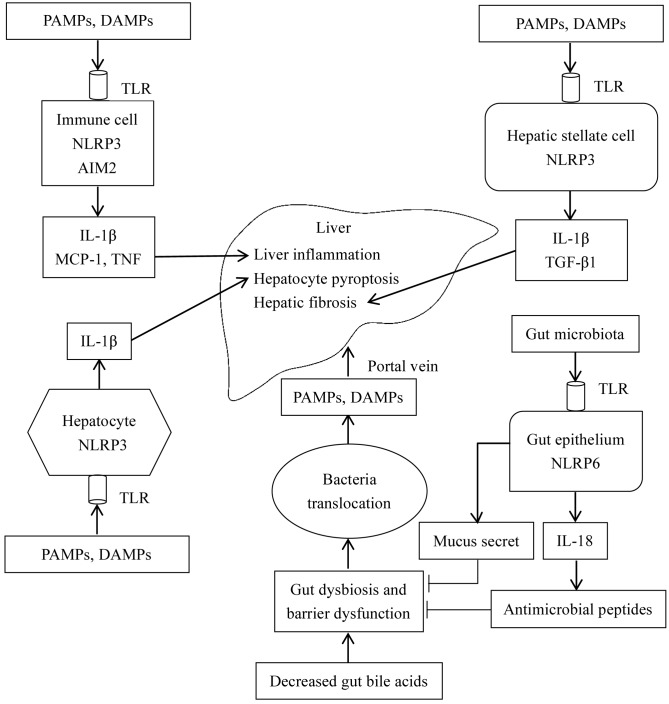
The gut‑liver axis connects the liver with the intestine via bile acid metabolism. Bile acid dysregulation leads to intestinal dysbiosis, that allows enterogenous pathogenic bacteria, including Gram‑negative bacteria and their products lipopolysaccharide (LPS), into the liver via the portal vein, triggering inflammation in the liver. The inflammasome serves as an intracellular pattern recognition receptor that detects pathogens or danger signals and mediates innate immunity in the liver or gut. Specifically, the NACHT, LRR and PYD domains‑containing protein (NLRP)6 inflammasome maintains intestinal microbial balance, by promoting interleukin (IL)‑18‑dependent antimicrobial peptide synthesis and mucus secretion from goblet cells. The NLRP3 inflammasome, in contrast, primarily induces IL‑1β and aggravates inflammatory liver injury. Furthermore, the NLRP3 inflammasome affects the epithelial integrity of cholangiocytes by inducing the production of pro‑inflammatory cytokines. In addition, bile acids, including deoxycholic acid and chenodeoxycholic acid, are able to activate the NLRP3 inflammasome in macrophages; however, bile acids have the potential to exert the opposite role by interacting with the membrane‑bound Takeda G‑protein receptor 5 or by activating nuclear farnesoid‑X receptor. Therefore, further investigation of the molecular mechanisms underlying the inflammasome, involved in the gut‑liver axis, may provide important insights into the identification of a potential therapeutic target for the treatment of liver and gut diseases. The present review discusses the roles of the inflammasome in the gut‑liver axis, and the emerging associations between the inflammasome and the intestinal microbiota or the bile acids in the gut‑liver axis.
Keywords: bile acid; gut-liver axis; inflammation; intestinal microbiota; NLRP3 inflammasome; NLRP6 inflammasome.
Figures
Similar articles
-
Gut-Liver Axis and Inflammasome Activation in Cholangiocyte Pathophysiology.Cells. 2020 Mar 17;9(3):736. doi: 10.3390/cells9030736. Cells. 2020. PMID: 32192118 Free PMC article. Review.
-
Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis.Oncotarget. 2016 Dec 20;7(51):83951-83963. doi: 10.18632/oncotarget.13796. Oncotarget. 2016. PMID: 27924062 Free PMC article.
-
Gut-liver axis, cirrhosis and portal hypertension: the chicken and the egg.Hepatol Int. 2018 Feb;12(Suppl 1):24-33. doi: 10.1007/s12072-017-9798-x. Epub 2017 May 26. Hepatol Int. 2018. PMID: 28550391 Free PMC article.
-
Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome.Immunity. 2016 Oct 18;45(4):802-816. doi: 10.1016/j.immuni.2016.09.008. Epub 2016 Sep 28. Immunity. 2016. PMID: 27692610
-
The NLRP6 inflammasome.Immunology. 2021 Mar;162(3):281-289. doi: 10.1111/imm.13293. Epub 2020 Dec 27. Immunology. 2021. PMID: 33314083 Free PMC article. Review.
Cited by
-
Primary biliary cholangitis: molecular pathogenesis perspectives and therapeutic potential of natural products.Front Immunol. 2023 Jun 30;14:1164202. doi: 10.3389/fimmu.2023.1164202. eCollection 2023. Front Immunol. 2023. PMID: 37457696 Free PMC article. Review.
-
Bile Acid Toxicity and Protein Kinases.Adv Exp Med Biol. 2021;1275:229-258. doi: 10.1007/978-3-030-49844-3_9. Adv Exp Med Biol. 2021. PMID: 33539018
-
Significance of elevated serum and hepatic NOD-like receptor pyrin domain containing 3 (NLRP3) in hepatitis C virus-related liver disease.Sci Rep. 2022 Nov 14;12(1):19528. doi: 10.1038/s41598-022-22022-5. Sci Rep. 2022. PMID: 36376416 Free PMC article.
-
Pattern Recognition Receptors: Significance of Expression in the Liver.Arch Immunol Ther Exp (Warsz). 2020 Sep 17;68(5):29. doi: 10.1007/s00005-020-00595-1. Arch Immunol Ther Exp (Warsz). 2020. PMID: 32944845 Free PMC article. Review.
-
Gut microbiota in health and disease: advances and future prospects.MedComm (2020). 2024 Nov 20;5(12):e70012. doi: 10.1002/mco2.70012. eCollection 2024 Dec. MedComm (2020). 2024. PMID: 39568773 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
