

Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes
- PMID: 30469440
- PMCID: PMC6266221
- DOI: 10.3390/genes9110566
Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes
Erratum in
-
Molecular Processes Connecting DNA Methylation Patterns with DNA Methyltransferases and Histone Modifications in Mammalian Genomes.Genes (Basel). 2019 May 21;10(5):388. doi: 10.3390/genes10050388. Genes (Basel). 2019. PMID: 31117298 Free PMC article.
Abstract
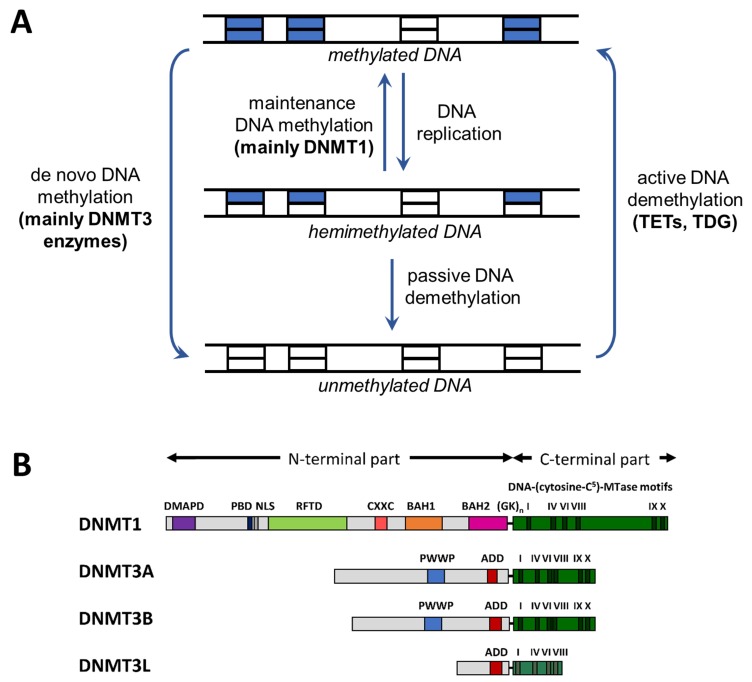
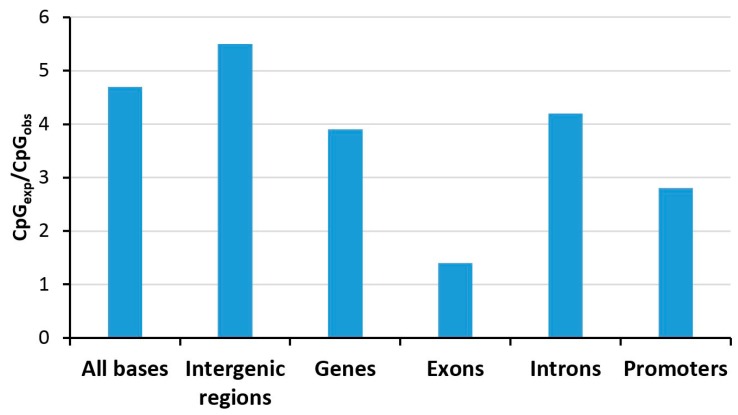
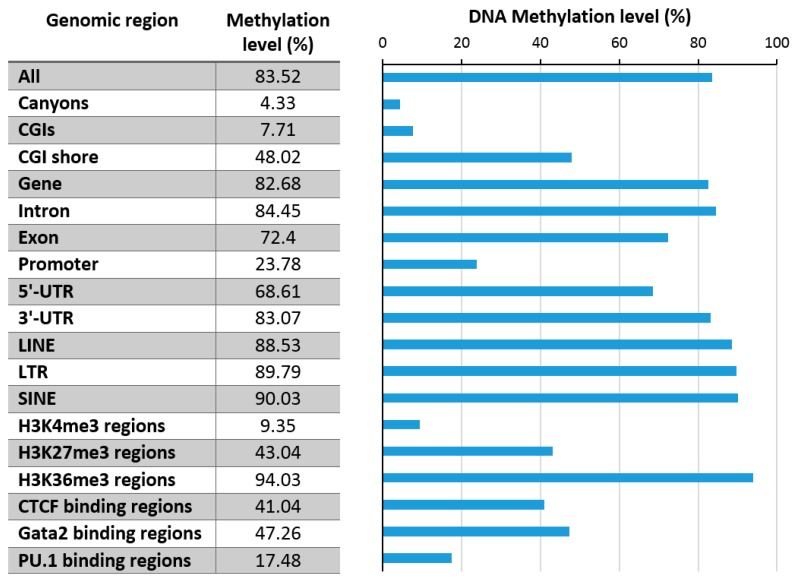
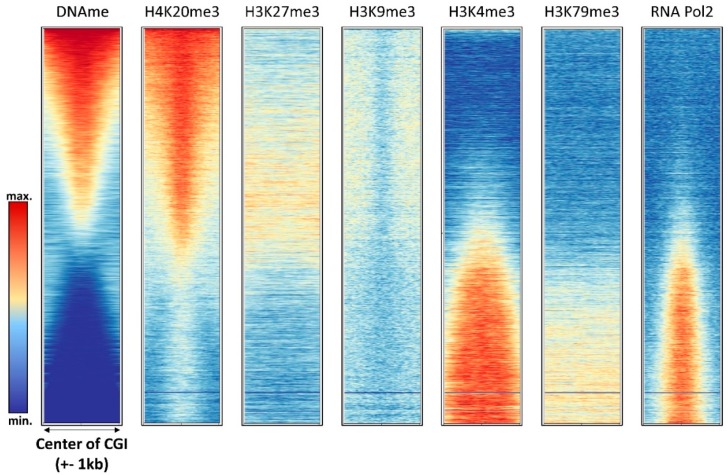
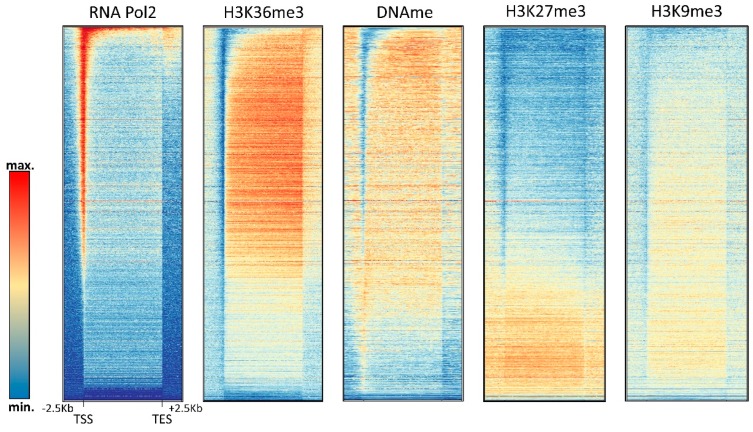
DNA methylation is an essential part of the epigenome chromatin modification network, which also comprises several covalent histone protein post-translational modifications. All these modifications are highly interconnected, because the writers and erasers of one mark, DNA methyltransferases (DNMTs) and ten eleven translocation enzymes (TETs) in the case of DNA methylation, are directly or indirectly targeted and regulated by other marks. Here, we have collected information about the genomic distribution and variability of DNA methylation in human and mouse DNA in different genomic elements. After summarizing the impact of DNA methylation on genome evolution including CpG depletion, we describe the connection of DNA methylation with several important histone post-translational modifications, including methylation of H3K4, H3K9, H3K27, and H3K36, but also with nucleosome remodeling. Moreover, we present the mechanistic features of mammalian DNA methyltransferases and their associated factors that mediate the crosstalk between DNA methylation and chromatin modifications. Finally, we describe recent advances regarding the methylation of non-CpG sites, methylation of adenine residues in human cells and methylation of mitochondrial DNA. At several places, we highlight controversial findings or open questions demanding future experimental work.
Keywords: DNA methylation; DNA methyltransferase; histone modification; molecular epigenetics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Editorial-Role of DNA Methyltransferases in the Epigenome.Genes (Basel). 2019 Jul 30;10(8):574. doi: 10.3390/genes10080574. Genes (Basel). 2019. PMID: 31366147 Free PMC article.
-
Profile of histone lysine methylation across transcribed mammalian chromatin.Mol Cell Biol. 2006 Dec;26(24):9185-95. doi: 10.1128/MCB.01529-06. Epub 2006 Oct 9. Mol Cell Biol. 2006. PMID: 17030614 Free PMC article.
-
DNMTs and Impact of CpG Content, Transcription Factors, Consensus Motifs, lncRNAs, and Histone Marks on DNA Methylation.Genes (Basel). 2020 Nov 12;11(11):1336. doi: 10.3390/genes11111336. Genes (Basel). 2020. PMID: 33198240 Free PMC article. Review.
-
Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together?J Cell Biochem. 2002;87(2):117-25. doi: 10.1002/jcb.10286. J Cell Biochem. 2002. PMID: 12244565 Review.
-
Histone Methylation by SET Domain Proteins in Fungi.Annu Rev Microbiol. 2017 Sep 8;71:413-439. doi: 10.1146/annurev-micro-102215-095757. Epub 2017 Jul 17. Annu Rev Microbiol. 2017. PMID: 28715960 Review.
Cited by
-
Locus-Specific and Stable DNA Demethylation at the H19/IGF2 ICR1 by Epigenome Editing Using a dCas9-SunTag System and the Catalytic Domain of TET1.Genes (Basel). 2024 Jan 8;15(1):80. doi: 10.3390/genes15010080. Genes (Basel). 2024. PMID: 38254969 Free PMC article.
-
DNA sequence-dependent activity and base flipping mechanisms of DNMT1 regulate genome-wide DNA methylation.Nat Commun. 2020 Jul 24;11(1):3723. doi: 10.1038/s41467-020-17531-8. Nat Commun. 2020. PMID: 32709850 Free PMC article.
-
Novel methylation mark and essential hypertension.J Genet Eng Biotechnol. 2022 Jan 21;20(1):11. doi: 10.1186/s43141-022-00301-y. J Genet Eng Biotechnol. 2022. PMID: 35061109 Free PMC article. Review.
-
Epigenetics in hepatocellular carcinoma development and therapy: The tip of the iceberg.JHEP Rep. 2020 Aug 7;2(6):100167. doi: 10.1016/j.jhepr.2020.100167. eCollection 2020 Dec. JHEP Rep. 2020. PMID: 33134907 Free PMC article. Review.
-
Abnormal Homocysteine Metabolism: An Insight of Alzheimer's Disease from DNA Methylation.Behav Neurol. 2020 Sep 8;2020:8438602. doi: 10.1155/2020/8438602. eCollection 2020. Behav Neurol. 2020. PMID: 32963633 Free PMC article. Review.
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
