

Dynamin 2 (DNM2) as Cause of, and Modifier for, Human Neuromuscular Disease
- PMID: 30426359
- PMCID: PMC6277281
- DOI: 10.1007/s13311-018-00686-0
Dynamin 2 (DNM2) as Cause of, and Modifier for, Human Neuromuscular Disease
Abstract
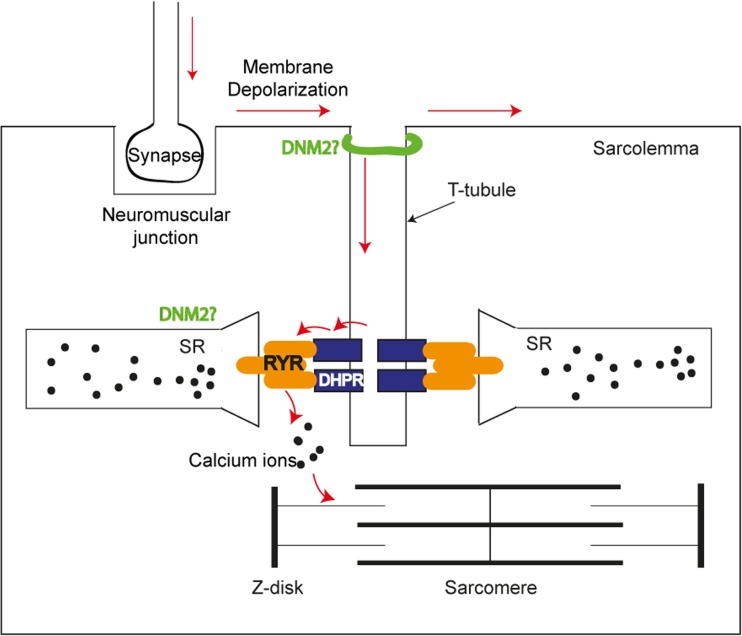
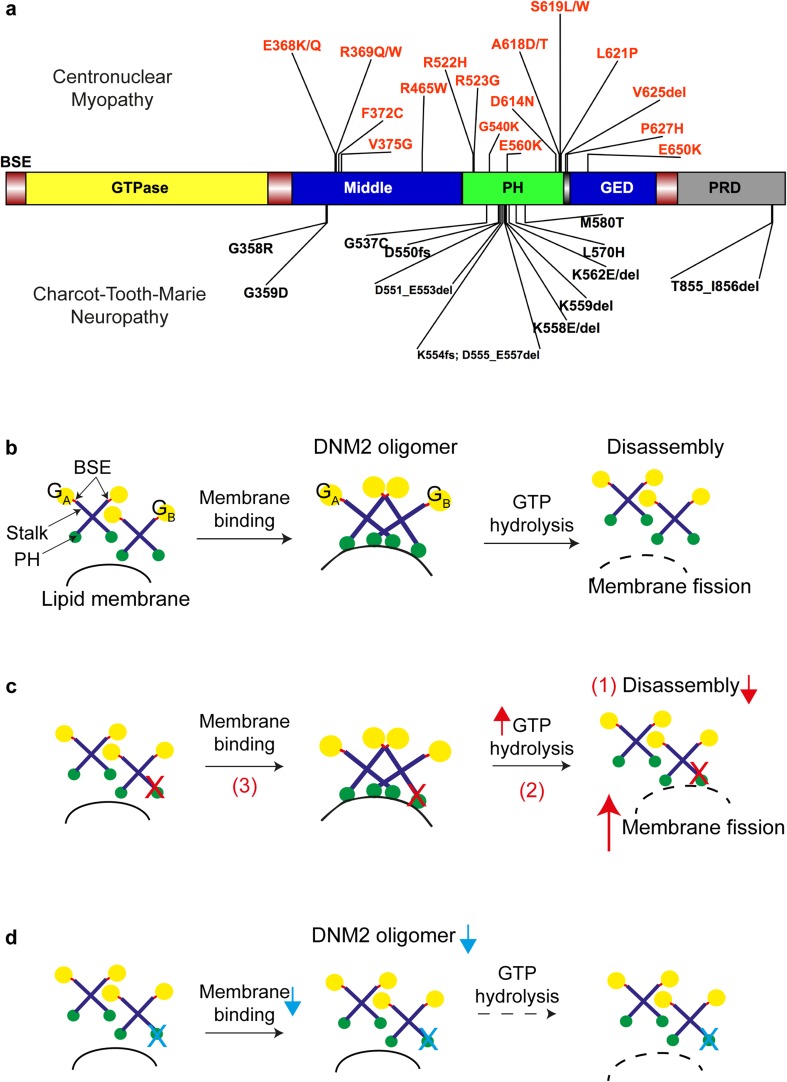
Dynamin 2 (DNM2) belongs to a family of large GTPases that are well known for mediating membrane fission by oligomerizing at the neck of membrane invaginations. Autosomal dominant mutations in the ubiquitously expressed DNM2 cause 2 discrete neuromuscular diseases: autosomal dominant centronuclear myopathy (ADCNM) and dominant intermediate Charcot-Marie-Tooth neuropathy (CMT). CNM and CMT mutations may affect DNM2 in distinct manners: CNM mutations may cause protein hyperactivity with elevated GTPase and fission activities, while CMT mutations could impair DNM2 lipid binding and activity. DNM2 is also a modifier of the X-linked and autosomal recessive forms of CNM, as DNM2 protein levels are upregulated in animal models and patient muscle samples. Strikingly, reducing DNM2 has been shown to revert muscle phenotypes in preclinical models of CNM. As DNM2 emerges as the key player in CNM pathogenesis, the role(s) of DNM2 in skeletal muscle remains unclear. This review aims to provide insights into potential pathomechanisms related to DNM2-CNM mutations, and discuss exciting outcomes of current and future therapeutic approaches targeting DNM2 hyperactivity.
Keywords: Centronuclear myopathy; Charcot–Marie–Tooth neuropathy; Congenital neuromuscular disorders; DNM2; Gene therapy.
Figures

Similar articles
-
Different in vivo impacts of dynamin 2 mutations implicated in Charcot-Marie-Tooth neuropathy or centronuclear myopathy.Hum Mol Genet. 2019 Dec 15;28(24):4067-4077. doi: 10.1093/hmg/ddz249. Hum Mol Genet. 2019. PMID: 31628461
-
Muscle-specific function of the centronuclear myopathy and Charcot-Marie-Tooth neuropathy-associated dynamin 2 is required for proper lipid metabolism, mitochondria, muscle fibers, neuromuscular junctions and peripheral nerves.Hum Mol Genet. 2013 Nov 1;22(21):4417-29. doi: 10.1093/hmg/ddt292. Epub 2013 Jun 27. Hum Mol Genet. 2013. PMID: 23813975
-
Insights into wild-type dynamin 2 and the consequences of DNM2 mutations from transgenic zebrafish.Hum Mol Genet. 2019 Dec 15;28(24):4186-4196. doi: 10.1093/hmg/ddz260. Hum Mol Genet. 2019. PMID: 31691805
-
A rare case of centronuclear myopathy with DNM2 mutation: genotype-phenotype correlation.Autops Case Rep. 2017 Jun 30;7(2):43-48. doi: 10.4322/acr.2017.020. eCollection 2017 Apr-Jun. Autops Case Rep. 2017. PMID: 28740838 Free PMC article. Review.
-
Dynamin 2 and human diseases.J Mol Med (Berl). 2010 Apr;88(4):339-50. doi: 10.1007/s00109-009-0587-4. Epub 2010 Feb 3. J Mol Med (Berl). 2010. PMID: 20127478 Review.
Cited by
-
Impairment of Autophagic Flux After Hypobaric Hypoxia Potentiates Oxidative Stress and Cognitive Function Disturbances in Mice.Neurosci Bull. 2024 Jan;40(1):35-49. doi: 10.1007/s12264-023-01099-6. Epub 2023 Aug 22. Neurosci Bull. 2024. PMID: 37608137 Free PMC article.
-
Scission, a critical step in autophagosome formation.Autophagy. 2020 Aug;16(8):1363-1365. doi: 10.1080/15548627.2020.1779468. Epub 2020 Jun 16. Autophagy. 2020. PMID: 32544363 Free PMC article.
-
Dynamin-Independent Mechanisms of Endocytosis and Receptor Trafficking.Cells. 2022 Aug 17;11(16):2557. doi: 10.3390/cells11162557. Cells. 2022. PMID: 36010634 Free PMC article. Review.
-
GSK3α phosphorylates dynamin-2 to promote GLUT4 endocytosis in muscle cells.J Cell Biol. 2023 Feb 6;222(2):e202102119. doi: 10.1083/jcb.202102119. Epub 2022 Nov 29. J Cell Biol. 2023. PMID: 36445308 Free PMC article.
-
Striated Preferentially Expressed Protein Kinase (SPEG) in Muscle Development, Function, and Disease.Int J Mol Sci. 2021 May 27;22(11):5732. doi: 10.3390/ijms22115732. Int J Mol Sci. 2021. PMID: 34072258 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
