

Activity of the SPCA1 Calcium Pump Couples Sphingomyelin Synthesis to Sorting of Secretory Proteins in the Trans-Golgi Network
- PMID: 30393074
- PMCID: PMC6261503
- DOI: 10.1016/j.devcel.2018.10.012
Activity of the SPCA1 Calcium Pump Couples Sphingomyelin Synthesis to Sorting of Secretory Proteins in the Trans-Golgi Network
Abstract
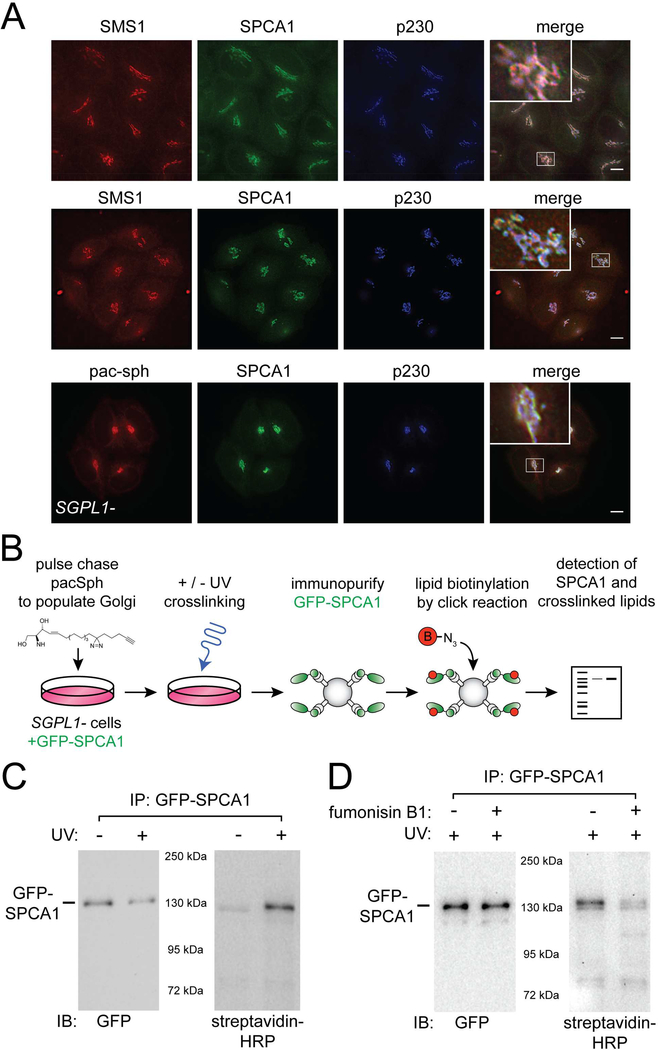
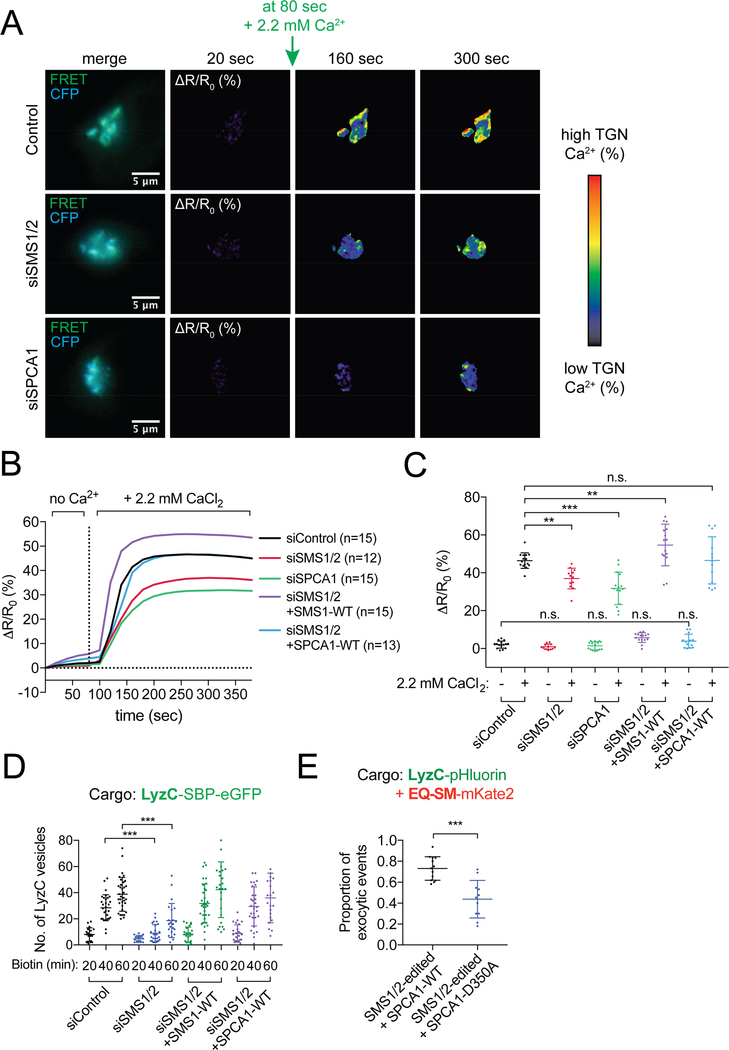
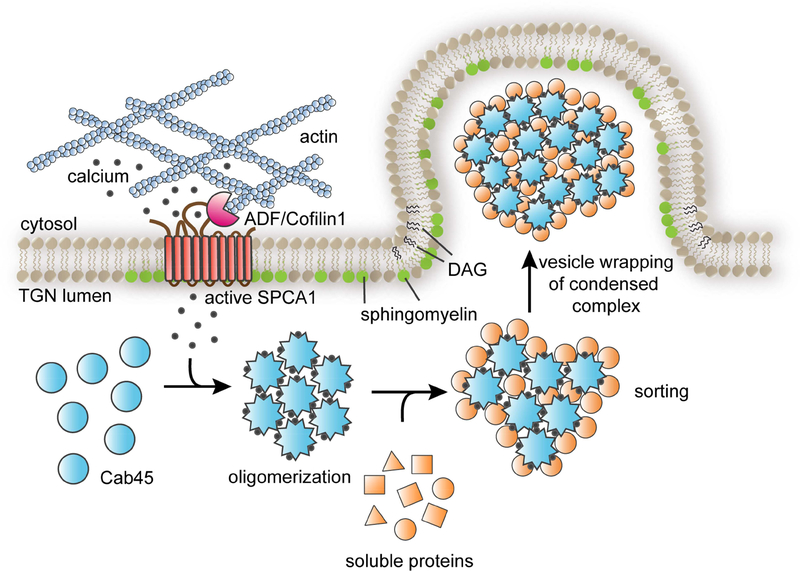
How the principal functions of the Golgi apparatus-protein processing, lipid synthesis, and sorting of macromolecules-are integrated to constitute cargo-specific trafficking pathways originating from the trans-Golgi network (TGN) is unknown. Here, we show that the activity of the Golgi localized SPCA1 calcium pump couples sorting and export of secreted proteins to synthesis of new lipid in the TGN membrane. A secreted Ca2+-binding protein, Cab45, constitutes the core component of a Ca2+-dependent, oligomerization-driven sorting mechanism whereby secreted proteins bound to Cab45 are packaged into a TGN-derived vesicular carrier whose membrane is enriched in sphingomyelin, a lipid implicated in TGN-to-cell surface transport. SPCA1 activity is controlled by the sphingomyelin content of the TGN membrane, such that local sphingomyelin synthesis promotes Ca2+ flux into the lumen of the TGN, which drives secretory protein sorting and export, thereby establishing a protein- and lipid-specific secretion pathway.
Keywords: Ca2+; Cab45; Cofilin; F-actin; Golgi apparatus; SPCA1; cargo sorting; secretion; sphingolipid metabolism; sphingomyelin.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interest:
The authors declare no competing interests.
Figures
Similar articles
-
Exploring new routes for secretory protein export from the trans-Golgi network.Mol Biol Cell. 2018 Feb 1;29(3):235-240. doi: 10.1091/mbc.E17-02-0117. Mol Biol Cell. 2018. PMID: 29382805 Free PMC article.
-
Secretory cargo sorting at the trans-Golgi network.Trends Cell Biol. 2014 Oct;24(10):584-93. doi: 10.1016/j.tcb.2014.04.007. Epub 2014 May 16. Trends Cell Biol. 2014. PMID: 24841758 Review.
-
ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1.Dev Cell. 2011 May 17;20(5):652-62. doi: 10.1016/j.devcel.2011.03.014. Dev Cell. 2011. PMID: 21571222
-
Secretory cargo sorting by Ca2+-dependent Cab45 oligomerization at the trans-Golgi network.J Cell Biol. 2016 May 9;213(3):305-14. doi: 10.1083/jcb.201601089. Epub 2016 May 2. J Cell Biol. 2016. PMID: 27138253 Free PMC article.
-
Cab45-Unraveling key features of a novel secretory cargo sorter at the trans-Golgi network.Eur J Cell Biol. 2017 Aug;96(5):383-390. doi: 10.1016/j.ejcb.2017.03.001. Epub 2017 Mar 18. Eur J Cell Biol. 2017. PMID: 28372832 Review.
Cited by
-
Lipid-dependent coupling of secretory cargo sorting and trafficking at the trans-Golgi network.FEBS Lett. 2019 Sep;593(17):2412-2427. doi: 10.1002/1873-3468.13552. Epub 2019 Jul 30. FEBS Lett. 2019. PMID: 31344259 Free PMC article. Review.
-
Nucleobindin-1 regulates ECM degradation by promoting intra-Golgi trafficking of MMPs.J Cell Biol. 2020 Aug 3;219(8):e201907058. doi: 10.1083/jcb.201907058. J Cell Biol. 2020. PMID: 32479594 Free PMC article.
-
The PKD-Dependent Biogenesis of TGN-to-Plasma Membrane Transport Carriers.Cells. 2021 Jun 28;10(7):1618. doi: 10.3390/cells10071618. Cells. 2021. PMID: 34203456 Free PMC article. Review.
-
Art2 mediates selective endocytosis of methionine transporters during adaptation to sphingolipid depletion.J Cell Sci. 2023 Jul 15;136(14):jcs260675. doi: 10.1242/jcs.260675. Epub 2023 Jul 25. J Cell Sci. 2023. PMID: 37337792 Free PMC article.
-
Comparative Proteomic Analysis of Pleurotus ostreatus Reveals Great Metabolic Differences in the Cap and Stipe Development and the Potential Role of Ca2+ in the Primordium Differentiation.Int J Mol Sci. 2019 Dec 14;20(24):6317. doi: 10.3390/ijms20246317. Int J Mol Sci. 2019. PMID: 31847351 Free PMC article.
References
-
- Ang SF, Fölsch H, 2012. The role of secretory and endocytic pathways in the maintenance of cell polarity. Essays Biochem. 53, 29–39. doi:10.1042/bse0530029 - DOI - PMC - PubMed
-
- Anitei M, Hoflack B, 2011. Exit from the trans-Golgi network: from molecules to mechanisms. Curr. Opin. Cell Biol 23, 443–451. doi:10.1016/j.ceb.2011.03.013 - DOI - PubMed
-
- Barenholz Y, Thompson TE, 1980. Sphingomyelins in bilayers and biological membranes. Biochim Biophys Acta 604, 129–158. - PubMed
-
- Baron CL, Malhotra V, 2002. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295, 325–328. doi:10.1126/science.1066759 - DOI - PubMed
-
- Baron S, Vangheluwe P, Sepúlveda MR, Wuytack F, Raeymaekers L, Vanoevelen J, 2010. The secretory pathway Ca(2+)-ATPase 1 is associated with cholesterol-rich microdomains of human colon adenocarcinoma cells. Biochim Biophys Acta 1798, 1512–1521. doi:10.1016/j.bbamem.2010.03.023 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
