

Temporal development of the gut microbiome in early childhood from the TEDDY study
- PMID: 30356187
- PMCID: PMC6415775
- DOI: 10.1038/s41586-018-0617-x
Temporal development of the gut microbiome in early childhood from the TEDDY study
Abstract
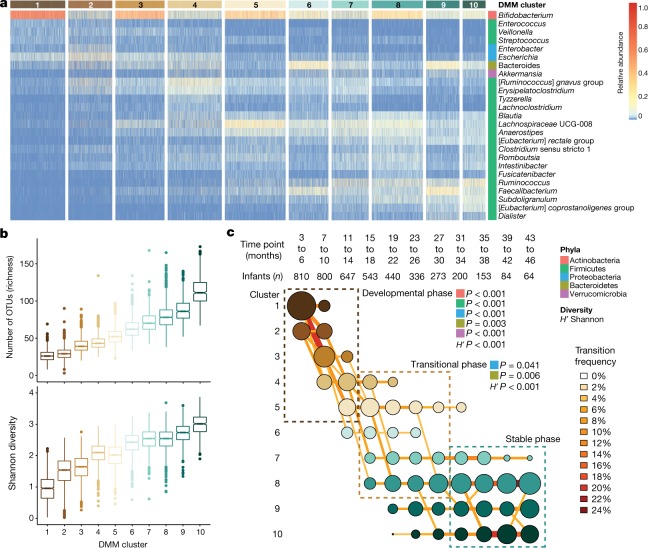
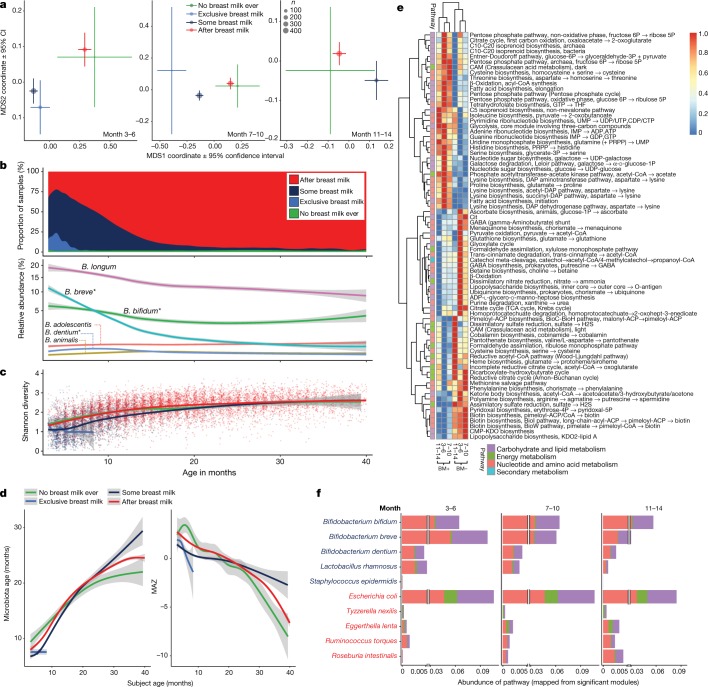
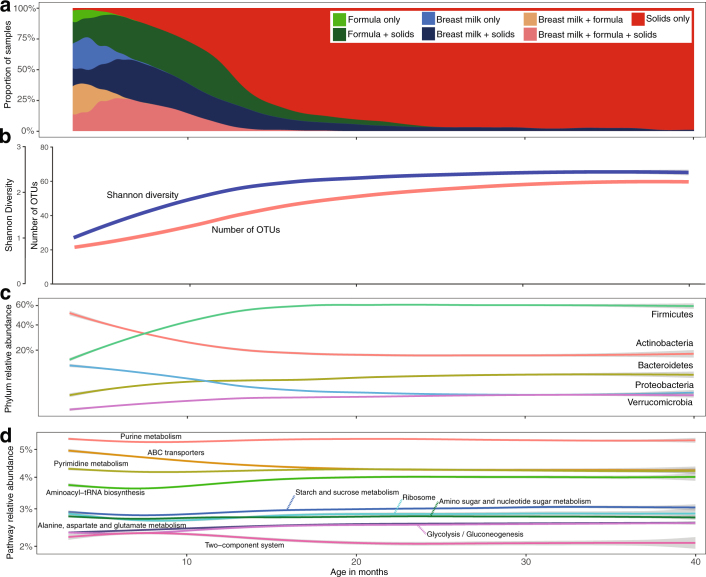
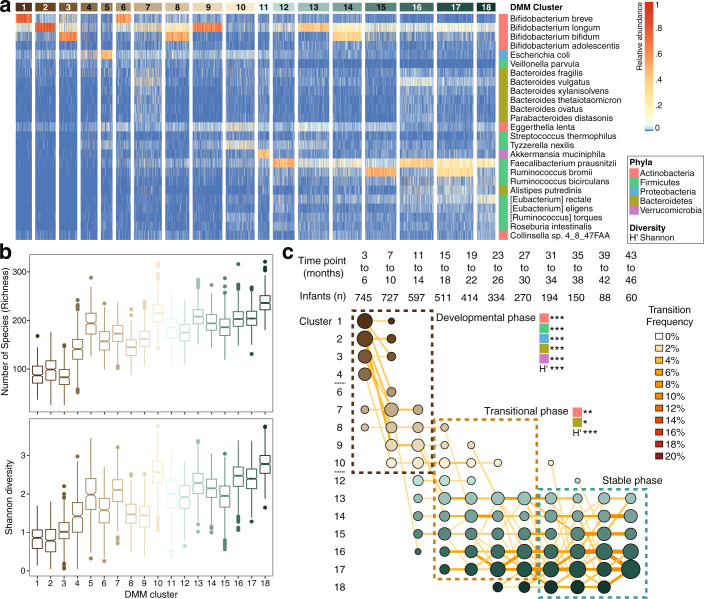
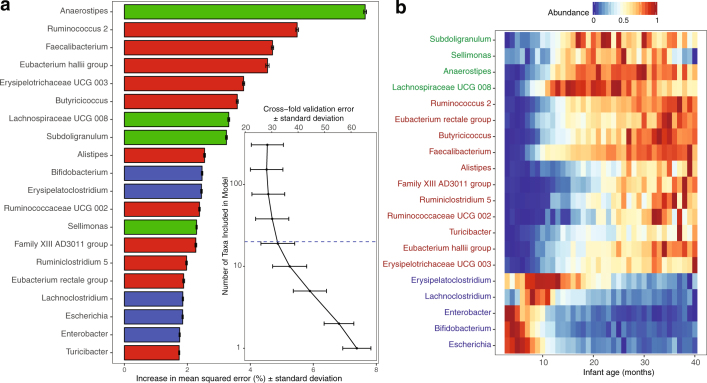
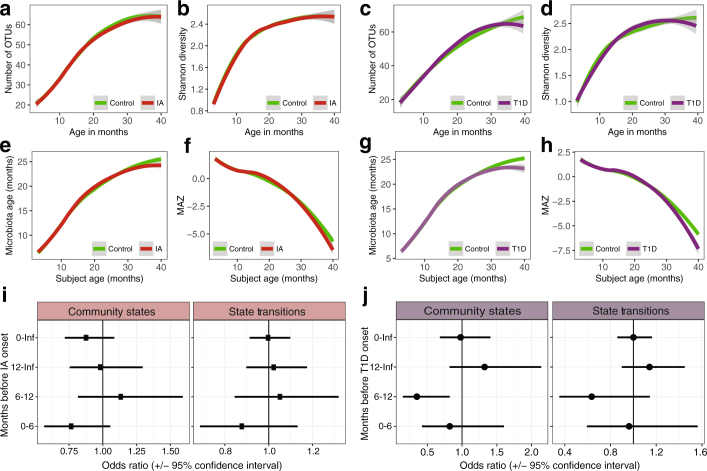
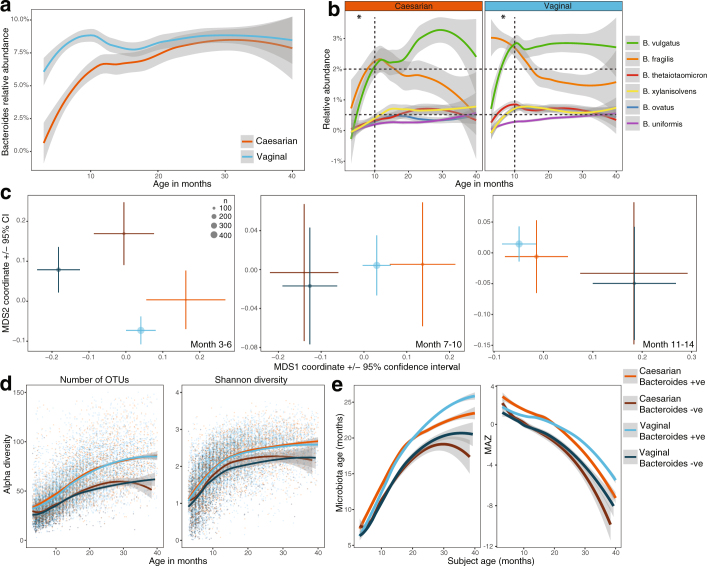
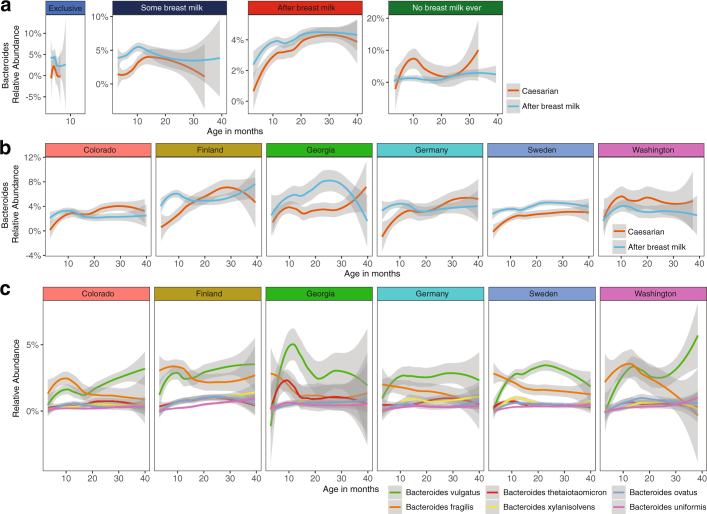
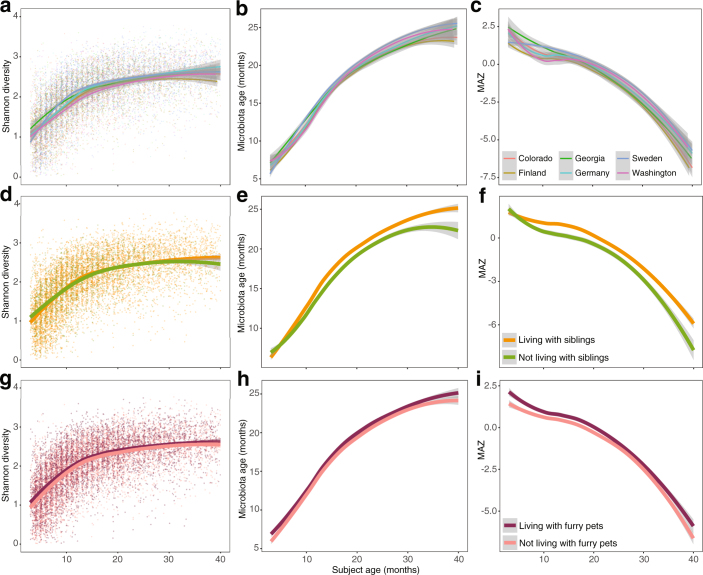
The development of the microbiome from infancy to childhood is dependent on a range of factors, with microbial-immune crosstalk during this time thought to be involved in the pathobiology of later life diseases1-9 such as persistent islet autoimmunity and type 1 diabetes10-12. However, to our knowledge, no studies have performed extensive characterization of the microbiome in early life in a large, multi-centre population. Here we analyse longitudinal stool samples from 903 children between 3 and 46 months of age by 16S rRNA gene sequencing (n = 12,005) and metagenomic sequencing (n = 10,867), as part of the The Environmental Determinants of Diabetes in the Young (TEDDY) study. We show that the developing gut microbiome undergoes three distinct phases of microbiome progression: a developmental phase (months 3-14), a transitional phase (months 15-30), and a stable phase (months 31-46). Receipt of breast milk, either exclusive or partial, was the most significant factor associated with the microbiome structure. Breastfeeding was associated with higher levels of Bifidobacterium species (B. breve and B. bifidum), and the cessation of breast milk resulted in faster maturation of the gut microbiome, as marked by the phylum Firmicutes. Birth mode was also significantly associated with the microbiome during the developmental phase, driven by higher levels of Bacteroides species (particularly B. fragilis) in infants delivered vaginally. Bacteroides was also associated with increased gut diversity and faster maturation, regardless of the birth mode. Environmental factors including geographical location and household exposures (such as siblings and furry pets) also represented important covariates. A nested case-control analysis revealed subtle associations between microbial taxonomy and the development of islet autoimmunity or type 1 diabetes. These data determine the structural and functional assembly of the microbiome in early life and provide a foundation for targeted mechanistic investigation into the consequences of microbial-immune crosstalk for long-term health.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
The human gut microbiome in early-onset type 1 diabetes from the TEDDY study.Nature. 2018 Oct;562(7728):589-594. doi: 10.1038/s41586-018-0620-2. Epub 2018 Oct 24. Nature. 2018. PMID: 30356183 Free PMC article.
-
Maternal diet during pregnancy is related with the infant stool microbiome in a delivery mode-dependent manner.Microbiome. 2018 Jul 5;6(1):109. doi: 10.1186/s40168-018-0490-8. Microbiome. 2018. PMID: 29973274 Free PMC article.
-
Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production.Microbiome. 2016 Apr 26;4:17. doi: 10.1186/s40168-016-0163-4. Microbiome. 2016. PMID: 27114075 Free PMC article.
-
The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants' life: a systematic review.BMC Gastroenterol. 2016 Jul 30;16(1):86. doi: 10.1186/s12876-016-0498-0. BMC Gastroenterol. 2016. PMID: 27475754 Free PMC article. Review.
-
Microbiome Composition in Pediatric Populations from Birth to Adolescence: Impact of Diet and Prebiotic and Probiotic Interventions.Dig Dis Sci. 2020 Mar;65(3):706-722. doi: 10.1007/s10620-020-06092-x. Dig Dis Sci. 2020. PMID: 32002758 Free PMC article. Review.
Cited by
-
Transcriptional networks in at-risk individuals identify signatures of type 1 diabetes progression.Sci Transl Med. 2021 Mar 31;13(587):eabd5666. doi: 10.1126/scitranslmed.abd5666. Sci Transl Med. 2021. PMID: 33790023 Free PMC article.
-
Early Onset of Autoimmune Diabetes in Children with Down Syndrome-Two Separate Aetiologies or an Immune System Pre-Programmed for Autoimmunity?Curr Diab Rep. 2020 Aug 25;20(9):47. doi: 10.1007/s11892-020-01318-8. Curr Diab Rep. 2020. PMID: 32839884 Free PMC article. Review.
-
Milk microbiome transplantation: recolonizing donor milk with mother's own milk microbiota.Appl Microbiol Biotechnol. 2024 Dec;108(1):74. doi: 10.1007/s00253-023-12965-8. Epub 2024 Jan 9. Appl Microbiol Biotechnol. 2024. PMID: 38194146 Free PMC article.
-
Food-breastmilk combinations alter the colonic microbiome of weaning infants: an in silico study.mSystems. 2024 Sep 17;9(9):e0057724. doi: 10.1128/msystems.00577-24. Epub 2024 Aug 27. mSystems. 2024. PMID: 39191378 Free PMC article.
-
The right educational environment: Oral tolerance in early life.Immunol Rev. 2024 Sep;326(1):17-34. doi: 10.1111/imr.13366. Epub 2024 Jul 13. Immunol Rev. 2024. PMID: 39001685 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK063821/DK/NIDDK NIH HHS/United States
- UC4 DK063863/DK/NIDDK NIH HHS/United States
- HHSN267200700014C/DK/NIDDK NIH HHS/United States
- U01 DK063861/DK/NIDDK NIH HHS/United States
- UL1 TR001427/TR/NCATS NIH HHS/United States
- U01 DK063790/DK/NIDDK NIH HHS/United States
- UL1 TR001082/TR/NCATS NIH HHS/United States
- UL1 TR000064/TR/NCATS NIH HHS/United States
- U01 DK063836/DK/NIDDK NIH HHS/United States
- U01 DK063829/DK/NIDDK NIH HHS/United States
- U01 DK063865/DK/NIDDK NIH HHS/United States
- UC4 DK095300/DK/NIDDK NIH HHS/United States
- UC4 DK063861/DK/NIDDK NIH HHS/United States
- UC4 DK063829/DK/NIDDK NIH HHS/United States
- UC4 DK063821/DK/NIDDK NIH HHS/United States
- UC4 DK117483/DK/NIDDK NIH HHS/United States
- UC4 DK063836/DK/NIDDK NIH HHS/United States
- UC4 DK112243/DK/NIDDK NIH HHS/United States
- UC4 DK063865/DK/NIDDK NIH HHS/United States
- U01 DK063863/DK/NIDDK NIH HHS/United States
- UC4 DK106955/DK/NIDDK NIH HHS/United States
- UC4 DK100238/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
