

A reference haplotype panel for genome-wide imputation of short tandem repeats
- PMID: 30353011
- PMCID: PMC6199332
- DOI: 10.1038/s41467-018-06694-0
A reference haplotype panel for genome-wide imputation of short tandem repeats
Abstract
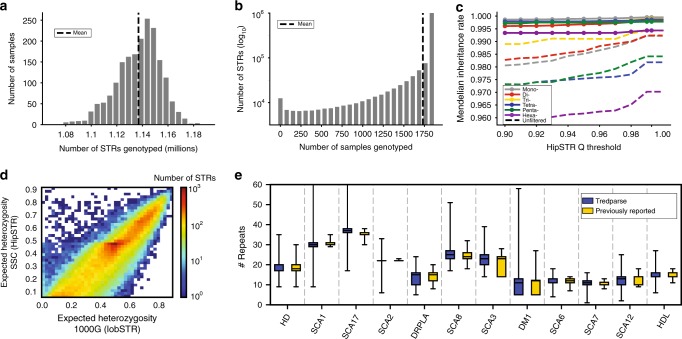
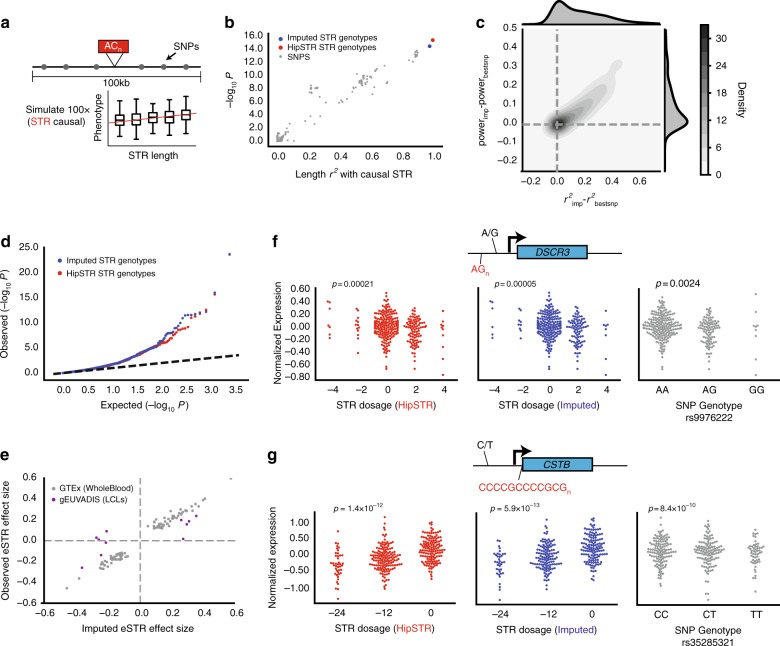
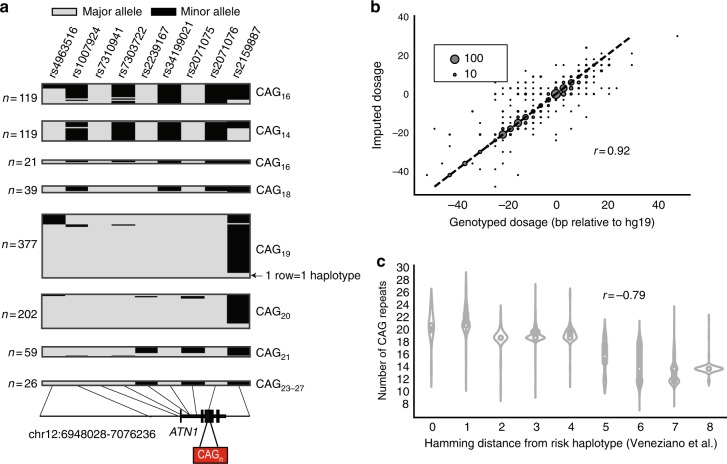
Short tandem repeats (STRs) are involved in dozens of Mendelian disorders and have been implicated in complex traits. However, genotyping arrays used in genome-wide association studies focus on single nucleotide polymorphisms (SNPs) and do not readily allow identification of STR associations. We leverage next-generation sequencing (NGS) from 479 families to create a SNP + STR reference haplotype panel. Our panel enables imputing STR genotypes into SNP array data when NGS is not available for directly genotyping STRs. Imputed genotypes achieve mean concordance of 97% with observed genotypes in an external dataset compared to 71% expected under a naive model. Performance varies widely across STRs, with near perfect concordance at bi-allelic STRs vs. 70% at highly polymorphic repeats. Imputation increases power over individual SNPs to detect STR associations with gene expression. Imputing STRs into existing SNP datasets will enable the first large-scale STR association studies across a range of complex traits.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Sequencing and characterizing short tandem repeats in the human genome.Nat Rev Genet. 2024 Jul;25(7):460-475. doi: 10.1038/s41576-024-00692-3. Epub 2024 Feb 16. Nat Rev Genet. 2024. PMID: 38366034 Review.
-
Evaluation of a SNP-STR haplotype panel for forensic genotype imputation.Forensic Sci Int Genet. 2023 Jan;62:102801. doi: 10.1016/j.fsigen.2022.102801. Epub 2022 Oct 17. Forensic Sci Int Genet. 2023. PMID: 36272212
-
Population-Scale Sequencing Data Enable Precise Estimates of Y-STR Mutation Rates.Am J Hum Genet. 2016 May 5;98(5):919-933. doi: 10.1016/j.ajhg.2016.04.001. Epub 2016 Apr 25. Am J Hum Genet. 2016. PMID: 27126583 Free PMC article.
-
Massive variation of short tandem repeats with functional consequences across strains of Arabidopsis thaliana.Genome Res. 2018 Aug;28(8):1169-1178. doi: 10.1101/gr.231753.117. Epub 2018 Jul 3. Genome Res. 2018. PMID: 29970452 Free PMC article.
-
On the Forensic Use of Y-Chromosome Polymorphisms.Genes (Basel). 2022 May 17;13(5):898. doi: 10.3390/genes13050898. Genes (Basel). 2022. PMID: 35627283 Free PMC article. Review.
Cited by
-
Analysis and benchmarking of small and large genomic variants across tandem repeats.Nat Biotechnol. 2024 Apr 26. doi: 10.1038/s41587-024-02225-z. Online ahead of print. Nat Biotechnol. 2024. PMID: 38671154
-
A comparison of software for analysis of rare and common short tandem repeat (STR) variation using human genome sequences from clinical and population-based samples.PLoS One. 2024 Apr 1;19(4):e0300545. doi: 10.1371/journal.pone.0300545. eCollection 2024. PLoS One. 2024. PMID: 38558075 Free PMC article.
-
Repeating themes of plastic genes and therapeutic schemes targeting the 'tandem repeatome'.Brain Commun. 2024 Feb 19;6(2):fcae047. doi: 10.1093/braincomms/fcae047. eCollection 2024. Brain Commun. 2024. PMID: 38449715 Free PMC article.
-
Modification of Huntington's disease by short tandem repeats.Brain Commun. 2024 Jan 23;6(2):fcae016. doi: 10.1093/braincomms/fcae016. eCollection 2024. Brain Commun. 2024. PMID: 38449714 Free PMC article.
-
Sequencing and characterizing short tandem repeats in the human genome.Nat Rev Genet. 2024 Jul;25(7):460-475. doi: 10.1038/s41576-024-00692-3. Epub 2024 Feb 16. Nat Rev Genet. 2024. PMID: 38366034 Review.
References
-
- Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ~700,000 individuals of European ancestry. Preprint at https://www.biorxiv.org/content/early/2018/03/22/274654 (2018). - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
