

Molecular pathways of nonalcoholic fatty liver disease development and progression
- PMID: 30343320
- PMCID: PMC11105781
- DOI: 10.1007/s00018-018-2947-0
Molecular pathways of nonalcoholic fatty liver disease development and progression
Abstract
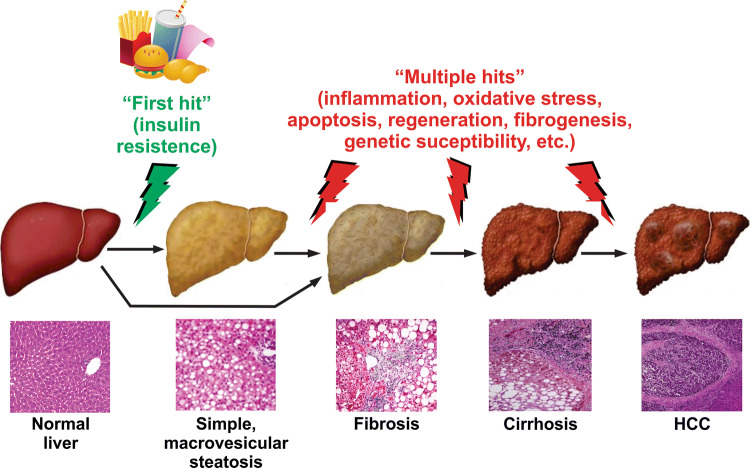
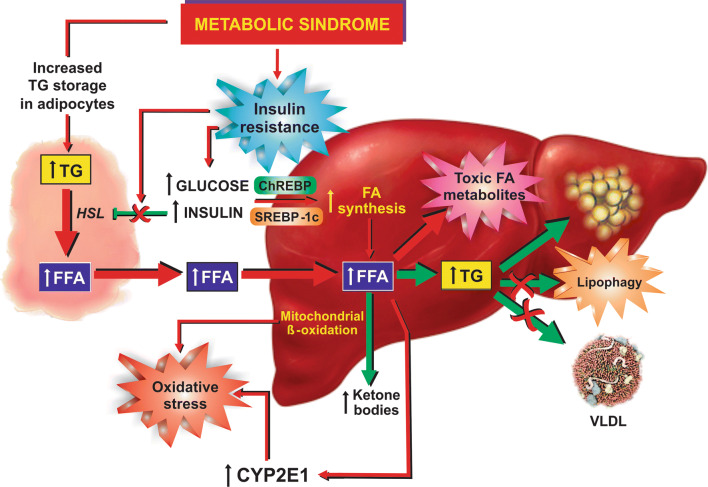
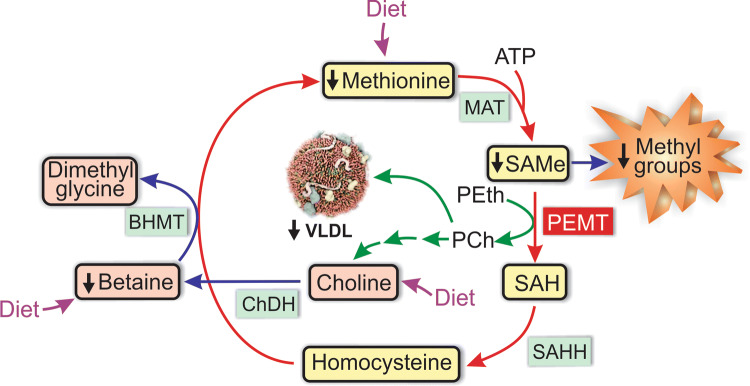
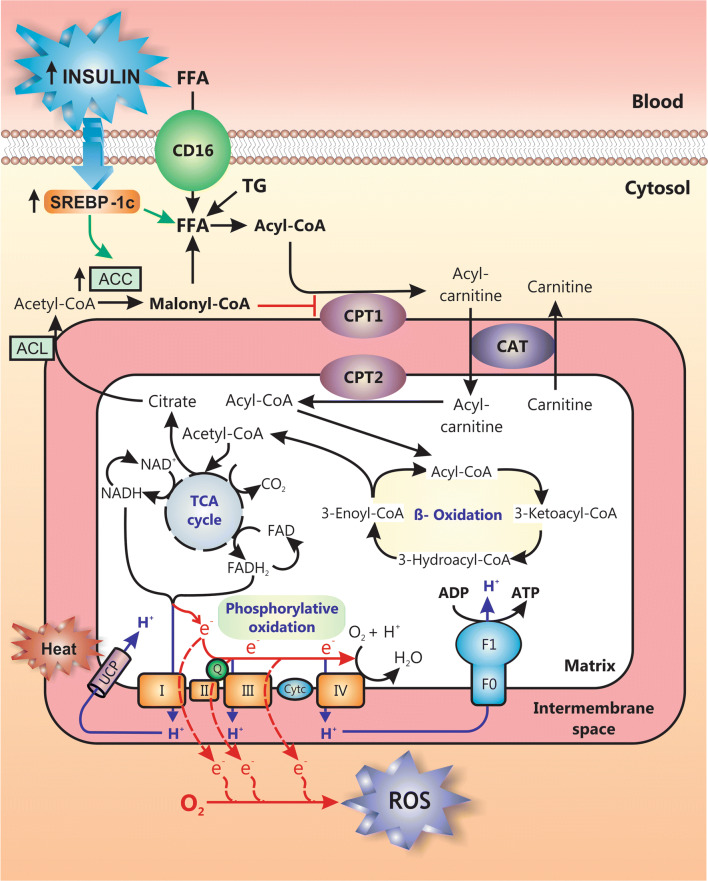
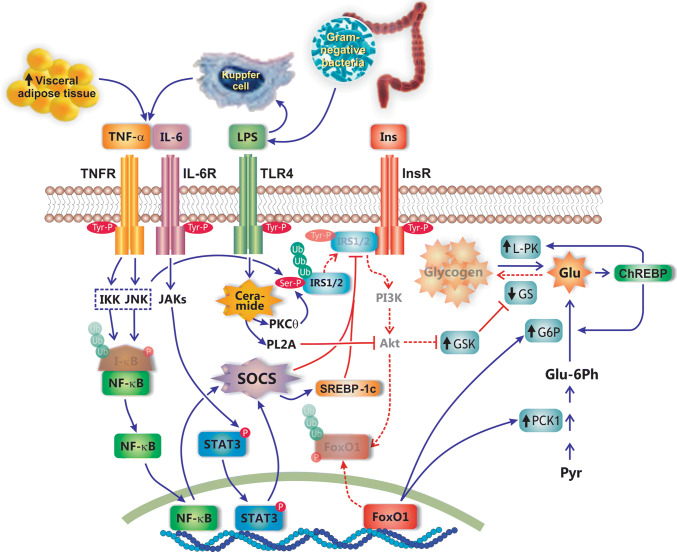
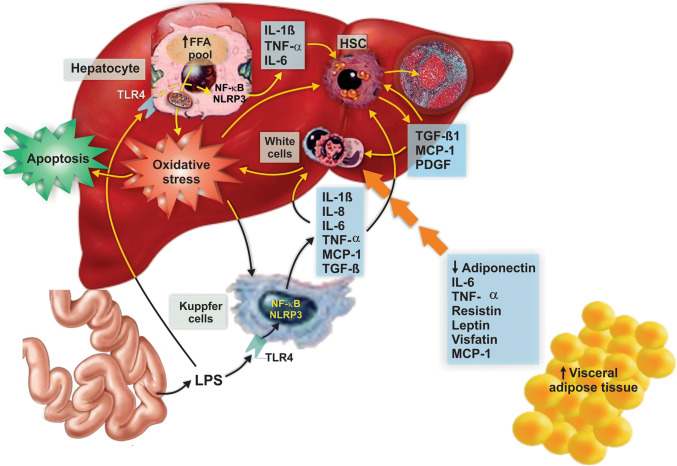
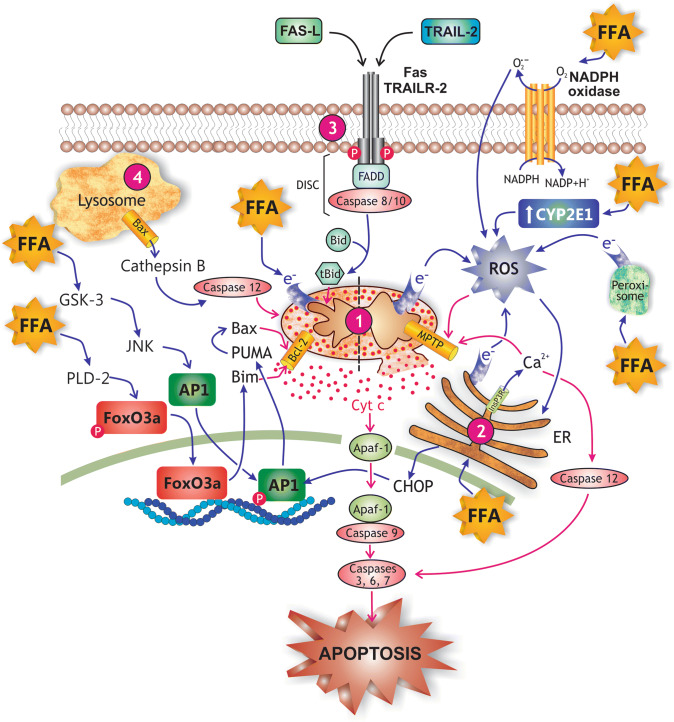
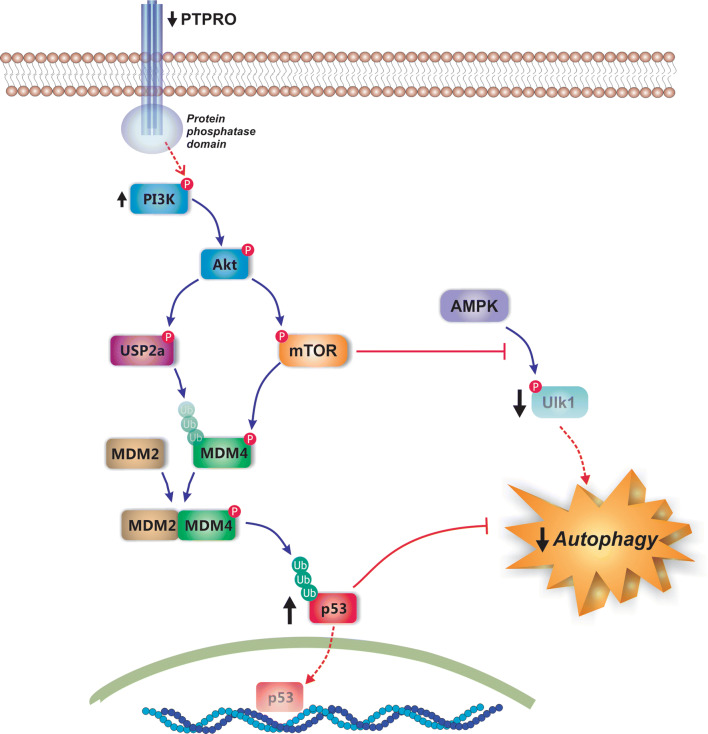
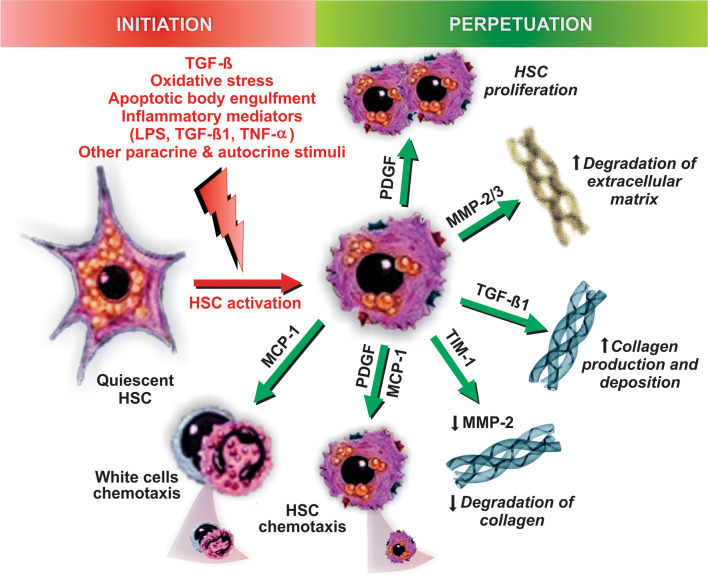
Nonalcoholic fatty liver disease (NAFLD) is a main hepatic manifestation of metabolic syndrome. It represents a wide spectrum of histopathological abnormalities ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) with or without fibrosis and, eventually, cirrhosis and hepatocellular carcinoma. While hepatic simple steatosis seems to be a rather benign manifestation of hepatic triglyceride accumulation, the buildup of highly toxic free fatty acids associated with insulin resistance-induced massive free fatty acid mobilization from adipose tissue and the increased de novo hepatic fatty acid synthesis from glucose acts as the "first hit" for NAFLD development. NAFLD progression seems to involve the occurrence of "parallel, multiple-hit" injuries, such as oxidative stress-induced mitochondrial dysfunction, endoplasmic reticulum stress, endotoxin-induced, TLR4-dependent release of inflammatory cytokines, and iron overload, among many others. These deleterious factors are responsible for the triggering of a number of signaling cascades leading to inflammation, cell death, and fibrosis, the hallmarks of NASH. This review is aimed at integrating the overwhelming progress made in the characterization of the physiopathological mechanisms of NAFLD at a molecular level, to better understand the factor influencing the initiation and progression of the disease.
Keywords: Hepatocellular death; Lipotoxicity; Liver fibrosis; Nonalcoholic fatty liver disease; Nonalcoholic steatohepatitis; Oxidative stress.
Figures
Similar articles
-
Nonalcoholic Fatty Liver Disease: Basic Pathogenetic Mechanisms in the Progression From NAFLD to NASH.Transplantation. 2019 Jan;103(1):e1-e13. doi: 10.1097/TP.0000000000002480. Transplantation. 2019. PMID: 30300287 Review.
-
New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis.Int J Mol Sci. 2018 Jul 13;19(7):2034. doi: 10.3390/ijms19072034. Int J Mol Sci. 2018. PMID: 30011790 Free PMC article. Review.
-
Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease.Oxid Med Cell Longev. 2018 Jun 11;2018:9547613. doi: 10.1155/2018/9547613. eCollection 2018. Oxid Med Cell Longev. 2018. PMID: 29991976 Free PMC article. Review.
-
Non-Alcoholic Fatty Liver Disease.Adv Exp Med Biol. 2017;960:443-467. doi: 10.1007/978-3-319-48382-5_19. Adv Exp Med Biol. 2017. PMID: 28585211 Review.
-
Fatty Acid and Glucose Sensors in Hepatic Lipid Metabolism: Implications in NAFLD.Semin Liver Dis. 2015 Aug;35(3):250-61. doi: 10.1055/s-0035-1562945. Epub 2015 Sep 17. Semin Liver Dis. 2015. PMID: 26378642 Review.
Cited by
-
Downregulation of hepatic lipopolysaccharide binding protein improves lipogenesis-induced liver lipid accumulation.Mol Ther Nucleic Acids. 2022 Aug 5;29:599-613. doi: 10.1016/j.omtn.2022.08.003. eCollection 2022 Sep 13. Mol Ther Nucleic Acids. 2022. PMID: 36090751 Free PMC article.
-
Adipocytokines: Are they the Theory of Everything?Cytokine. 2020 Sep;133:155144. doi: 10.1016/j.cyto.2020.155144. Epub 2020 Jun 16. Cytokine. 2020. PMID: 32559663 Free PMC article. Review.
-
Potential Therapeutic Implication of Herbal Medicine in Mitochondria-Mediated Oxidative Stress-Related Liver Diseases.Antioxidants (Basel). 2022 Oct 17;11(10):2041. doi: 10.3390/antiox11102041. Antioxidants (Basel). 2022. PMID: 36290765 Free PMC article. Review.
-
Oral Administration of Recombinant Lactoferrin-Expressing Probiotics Ameliorates Diet-Induced Lipid Accumulation and Inflammation in Non-Alcoholic Fatty Liver Disease in Mice.Microorganisms. 2022 Nov 9;10(11):2215. doi: 10.3390/microorganisms10112215. Microorganisms. 2022. PMID: 36363807 Free PMC article.
-
Mismatched effects of receptor interacting protein kinase-3 on hepatic steatosis and inflammation in non-alcoholic fatty liver disease.World J Gastroenterol. 2018 Dec 28;24(48):5477-5490. doi: 10.3748/wjg.v24.i48.5477. World J Gastroenterol. 2018. PMID: 30622377 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
