

Predicting microRNA targeting efficacy in Drosophila
- PMID: 30286781
- PMCID: PMC6172730
- DOI: 10.1186/s13059-018-1504-3
Predicting microRNA targeting efficacy in Drosophila
Abstract
Background: MicroRNAs (miRNAs) are short regulatory RNAs that derive from hairpin precursors. Important for understanding the functional roles of miRNAs is the ability to predict the messenger RNA (mRNA) targets most responsive to each miRNA. Progress towards developing quantitative models of miRNA targeting in Drosophila and other invertebrate species has lagged behind that of mammals due to the paucity of datasets measuring the effects of miRNAs on mRNA levels.
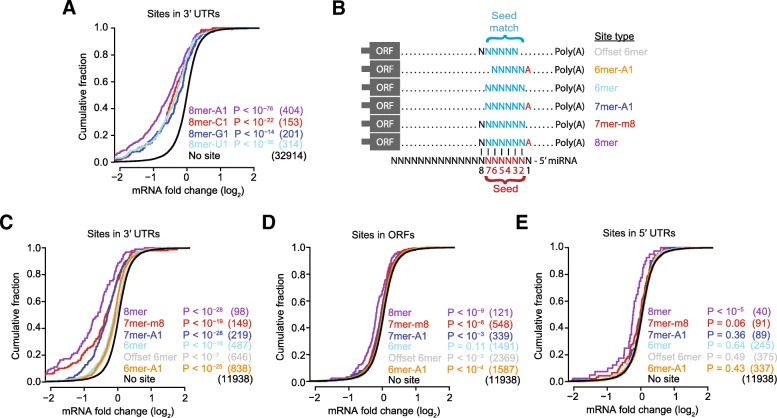
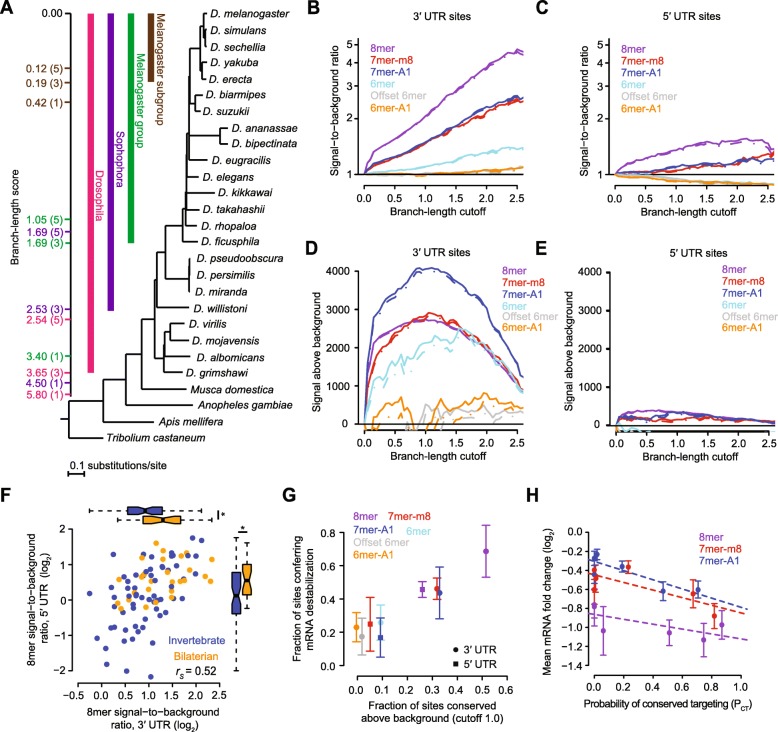
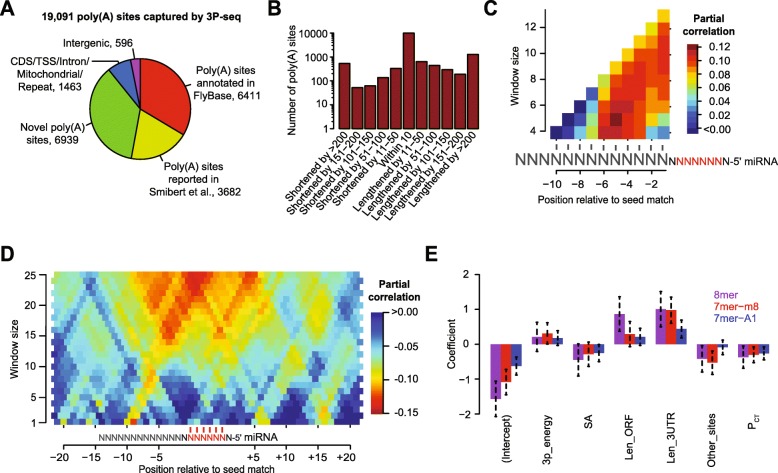
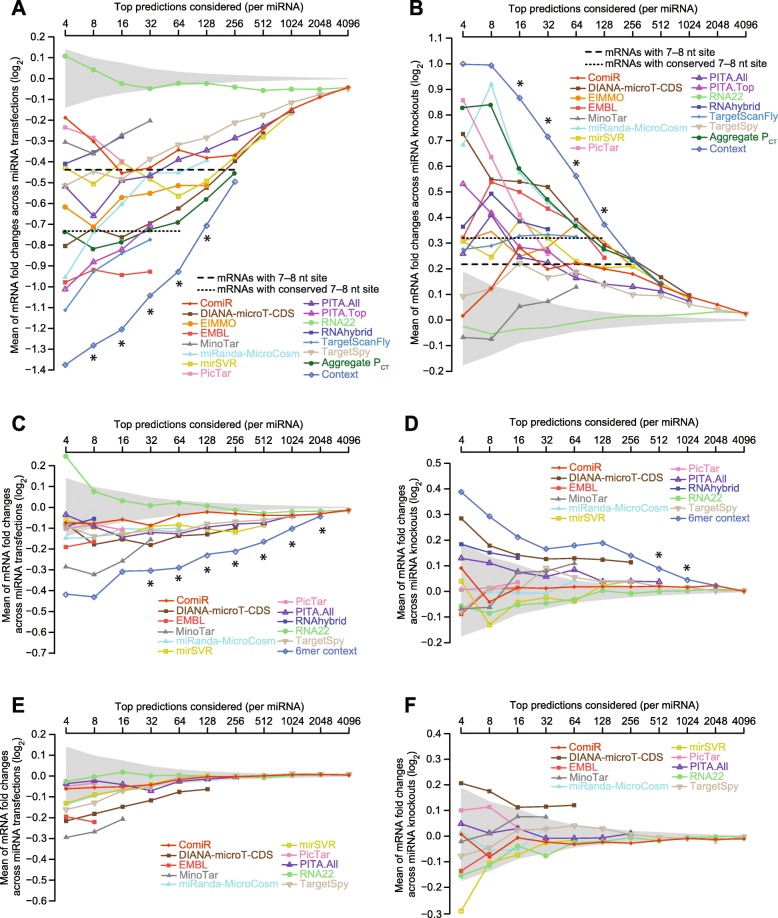
Results: We acquired datasets suitable for the quantitative study of miRNA targeting in Drosophila. Analyses of these data expanded the types of regulatory sites known to be effective in flies, expanded the mRNA regions with detectable targeting to include 5' untranslated regions, and identified features of site context that correlate with targeting efficacy in fly cells. Updated evolutionary analyses evaluated the probability of conserved targeting for each predicted site and indicated that more than a third of the Drosophila genes are preferentially conserved targets of miRNAs. Based on these results, a quantitative model was developed to predict targeting efficacy in insects. This model performed better than existing models, and it drives the most recent version, v7, of TargetScanFly.
Conclusions: Our evolutionary and functional analyses expand the known scope of miRNA targeting in flies and other insects. The existence of a quantitative model that has been developed and trained using Drosophila data will provide a valuable resource for placing miRNAs into gene regulatory networks of this important experimental organism.
Keywords: Non-coding RNAs; Post-transcriptional gene regulation; miRNA target prediction.
Conflict of interest statement
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Evolution of Hox post-transcriptional regulation by alternative polyadenylation and microRNA modulation within 12 Drosophila genomes.Mol Biol Evol. 2011 Sep;28(9):2453-60. doi: 10.1093/molbev/msr073. Epub 2011 Mar 24. Mol Biol Evol. 2011. PMID: 21436120
-
Analysis of microRNA-target interactions by a target structure based hybridization model.Pac Symp Biocomput. 2008:64-74. Pac Symp Biocomput. 2008. PMID: 18232104
-
Identifications of conserved 7-mers in 3'-UTRs and microRNAs in Drosophila.BMC Bioinformatics. 2007 Nov 8;8:432. doi: 10.1186/1471-2105-8-432. BMC Bioinformatics. 2007. PMID: 17996040 Free PMC article.
-
Small but influential: the role of microRNAs on gene regulatory network and 3'UTR evolution.J Genet Genomics. 2009 Jan;36(1):1-6. doi: 10.1016/S1673-8527(09)60001-1. J Genet Genomics. 2009. PMID: 19161940 Review.
-
Identifying miRNAs, targets and functions.Brief Bioinform. 2014 Jan;15(1):1-19. doi: 10.1093/bib/bbs075. Epub 2012 Nov 22. Brief Bioinform. 2014. PMID: 23175680 Free PMC article. Review.
Cited by
-
Dysregulated miRNA and mRNA Expression Affect Overlapping Pathways in a Huntington's Disease Model.Int J Mol Sci. 2023 Jul 26;24(15):11942. doi: 10.3390/ijms241511942. Int J Mol Sci. 2023. PMID: 37569316 Free PMC article.
-
Comprehensive analysis of circRNAs from cashmere goat skin by next generation RNA sequencing (RNA-seq).Sci Rep. 2020 Jan 16;10(1):516. doi: 10.1038/s41598-019-57404-9. Sci Rep. 2020. PMID: 31949277 Free PMC article.
-
Sites of transcription initiation drive mRNA isoform selection.Cell. 2023 May 25;186(11):2438-2455.e22. doi: 10.1016/j.cell.2023.04.012. Epub 2023 May 12. Cell. 2023. PMID: 37178687 Free PMC article.
-
Genome-wide kinetic profiling of pre-mRNA 3' end cleavage.RNA. 2024 Feb 16;30(3):256-270. doi: 10.1261/rna.079783.123. RNA. 2024. PMID: 38164598 Free PMC article.
-
Noncoding RNA Regulation of Hormonal and Metabolic Systems in the Fruit Fly Drosophila.Metabolites. 2023 Jan 19;13(2):152. doi: 10.3390/metabo13020152. Metabolites. 2023. PMID: 36837772 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
