

ChromTime: modeling spatio-temporal dynamics of chromatin marks
- PMID: 30097049
- PMCID: PMC6085762
- DOI: 10.1186/s13059-018-1485-2
ChromTime: modeling spatio-temporal dynamics of chromatin marks
Abstract
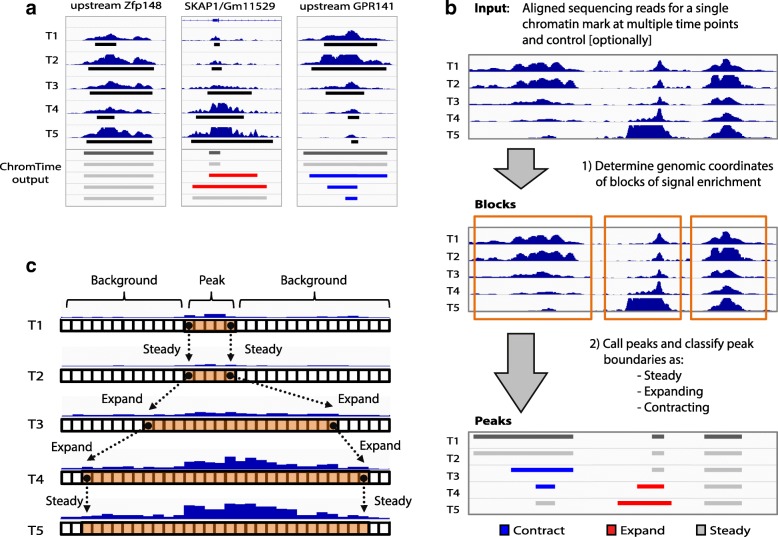
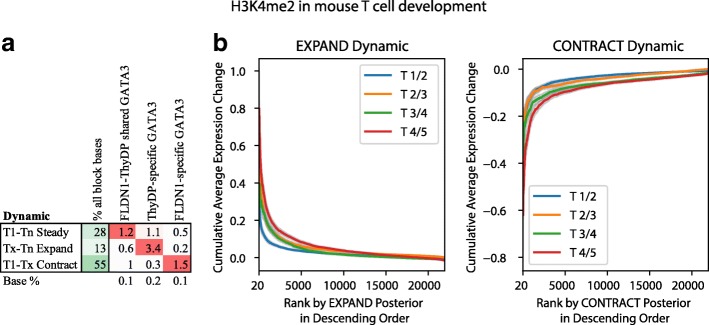
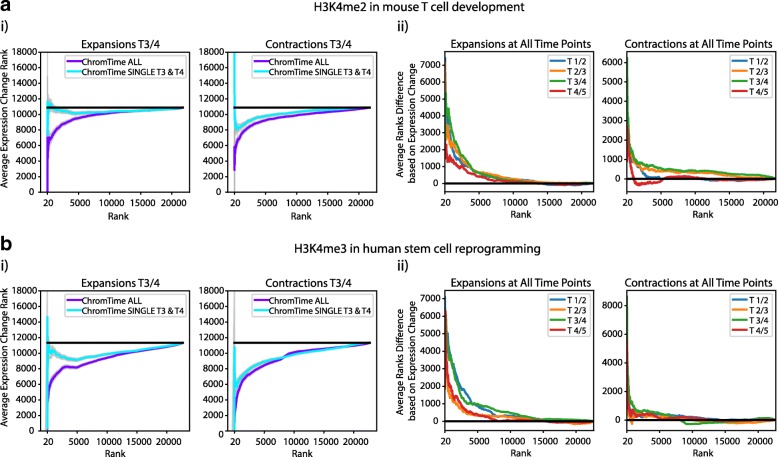
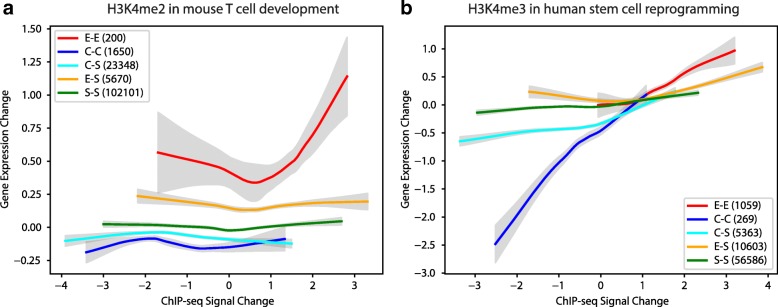
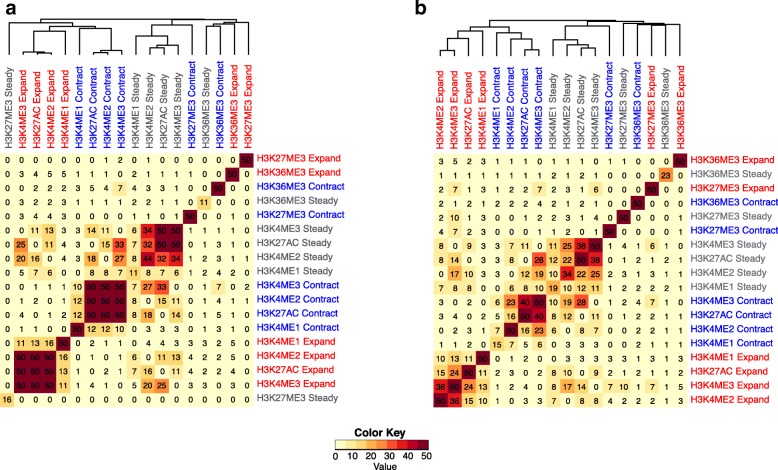
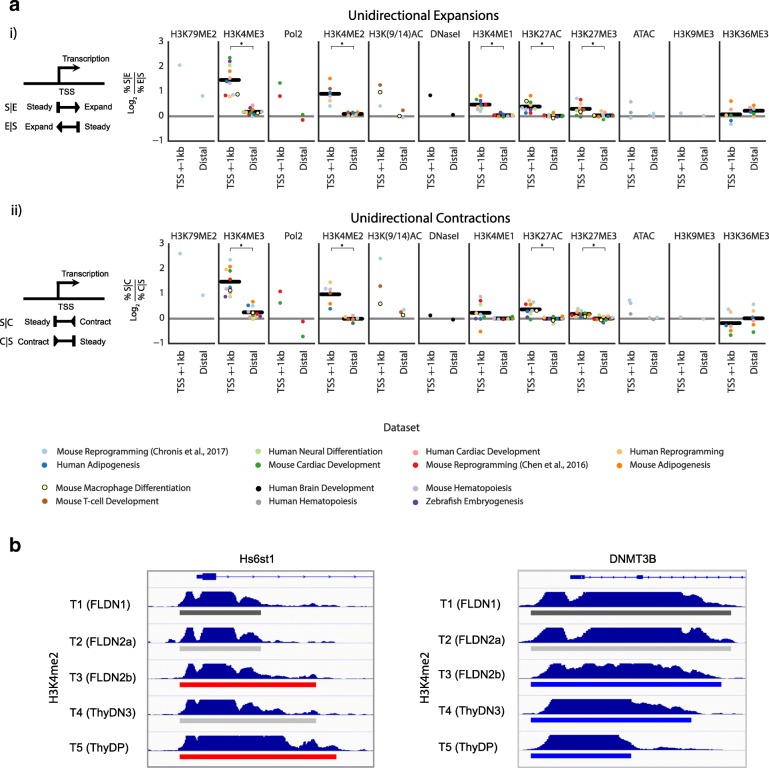
To model spatial changes of chromatin mark peaks over time we develop and apply ChromTime, a computational method that predicts peaks to be either expanding, contracting, or holding steady between time points. Predicted expanding and contracting peaks can mark regulatory regions associated with transcription factor binding and gene expression changes. Spatial dynamics of peaks provide information about gene expression changes beyond localized signal density changes. ChromTime detects asymmetric expansions and contractions, which for some marks associate with the direction of transcription. ChromTime facilitates the analysis of time course chromatin data in a range of biological systems.
Keywords: Chromatin marks; Epigenomics; Histone modifications; Spatial dynamics; Time course.
Conflict of interest statement
Not applicable.
Not applicable.
The authors declare that they have no competing interests.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

Similar articles
-
'Traffic light rules': Chromatin states direct miRNA-mediated network motifs running by integrating epigenome and regulatome.Biochim Biophys Acta. 2016 Jul;1860(7):1475-88. doi: 10.1016/j.bbagen.2016.04.008. Epub 2016 Apr 14. Biochim Biophys Acta. 2016. PMID: 27091612
-
Regulation of transcription of the steroidogenic acute regulatory protein (StAR) gene: temporal and spatial changes in transcription factor binding and histone modification.Mol Cell Endocrinol. 2004 Feb 27;215(1-2):119-26. doi: 10.1016/j.mce.2003.11.014. Mol Cell Endocrinol. 2004. PMID: 15026184
-
Genome-Wide Analysis of the Distinct Types of Chromatin Interactions in Arabidopsis thaliana.Plant Cell Physiol. 2017 Jan 1;58(1):57-70. doi: 10.1093/pcp/pcw194. Plant Cell Physiol. 2017. PMID: 28064247
-
Fast signals and slow marks: the dynamics of histone modifications.Trends Biochem Sci. 2010 Nov;35(11):618-26. doi: 10.1016/j.tibs.2010.05.006. Epub 2010 Aug 3. Trends Biochem Sci. 2010. PMID: 20685123 Review.
-
Regulation of alternative splicing through coupling with transcription and chromatin structure.Annu Rev Biochem. 2015;84:165-98. doi: 10.1146/annurev-biochem-060614-034242. Annu Rev Biochem. 2015. PMID: 26034889 Review.
Cited by
-
Nucleome programming is required for the foundation of totipotency in mammalian germline development.EMBO J. 2022 Jul 4;41(13):e110600. doi: 10.15252/embj.2022110600. Epub 2022 Jun 15. EMBO J. 2022. PMID: 35703121 Free PMC article.
-
From Genotype to Phenotype: Through Chromatin.Genes (Basel). 2019 Jan 23;10(2):76. doi: 10.3390/genes10020076. Genes (Basel). 2019. PMID: 30678090 Free PMC article. Review.
-
Histone H3K4me3 breadth in hypoxia reveals endometrial core functions and stress adaptation linked to endometriosis.iScience. 2022 Apr 12;25(5):104235. doi: 10.1016/j.isci.2022.104235. eCollection 2022 May 20. iScience. 2022. PMID: 35494227 Free PMC article.
-
Temporal Dynamic Methods for Bulk RNA-Seq Time Series Data.Genes (Basel). 2021 Feb 27;12(3):352. doi: 10.3390/genes12030352. Genes (Basel). 2021. PMID: 33673721 Free PMC article. Review.
-
Schizophrenia genomics: genetic complexity and functional insights.Nat Rev Neurosci. 2024 Sep;25(9):611-624. doi: 10.1038/s41583-024-00837-7. Epub 2024 Jul 19. Nat Rev Neurosci. 2024. PMID: 39030273 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
