

Lateral Gene Transfer Between Protozoa-Related Giant Viruses of Family Mimiviridae and Chlamydiae
- PMID: 30038484
- PMCID: PMC6050620
- DOI: 10.1177/1176934318788337
Lateral Gene Transfer Between Protozoa-Related Giant Viruses of Family Mimiviridae and Chlamydiae
Abstract
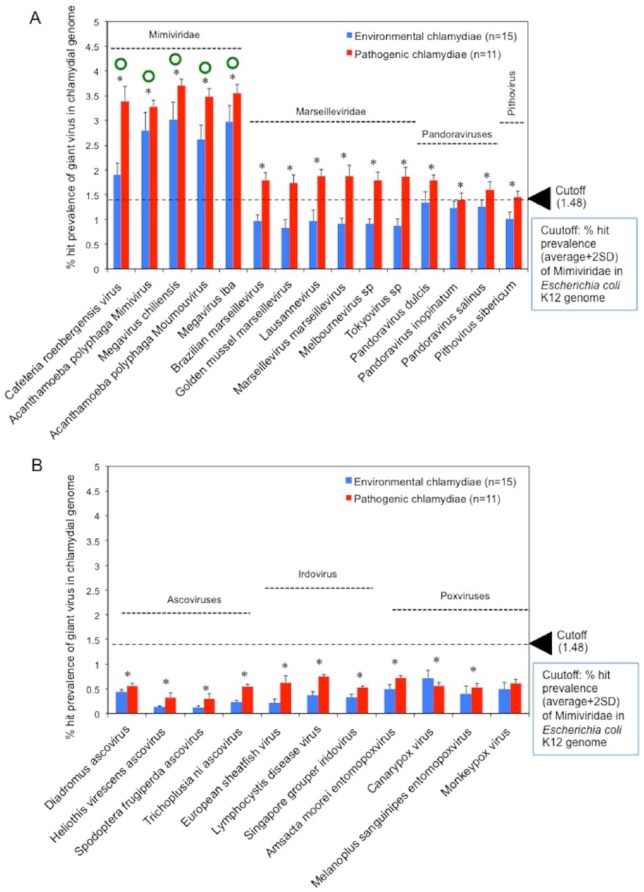
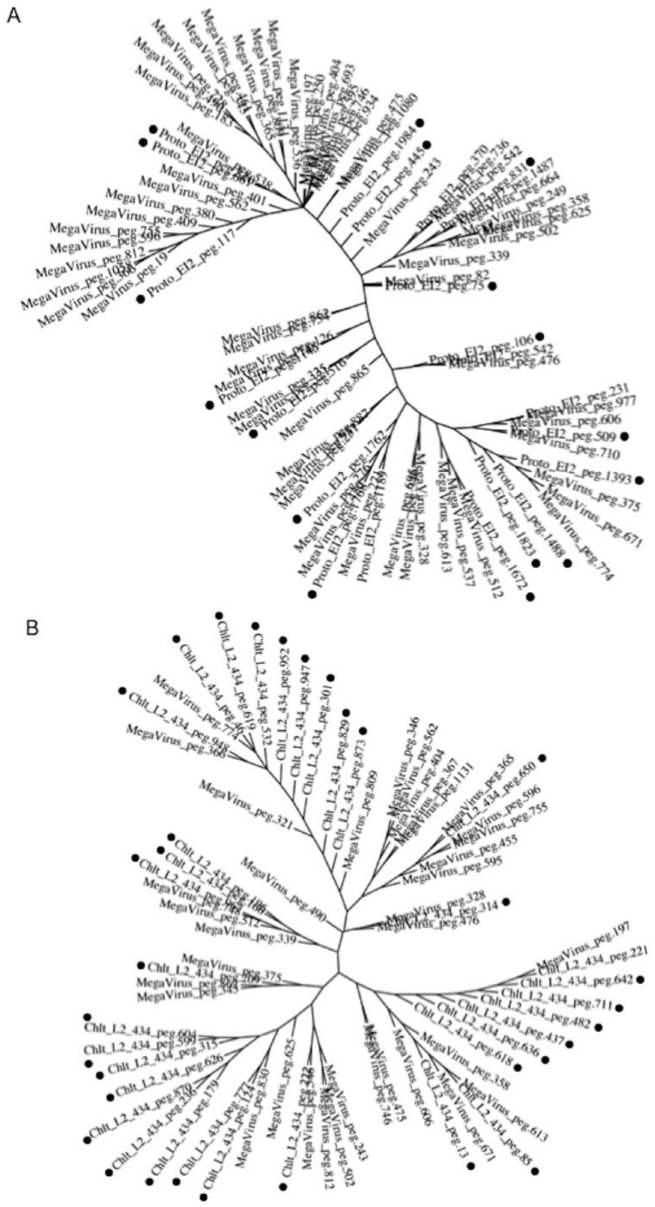
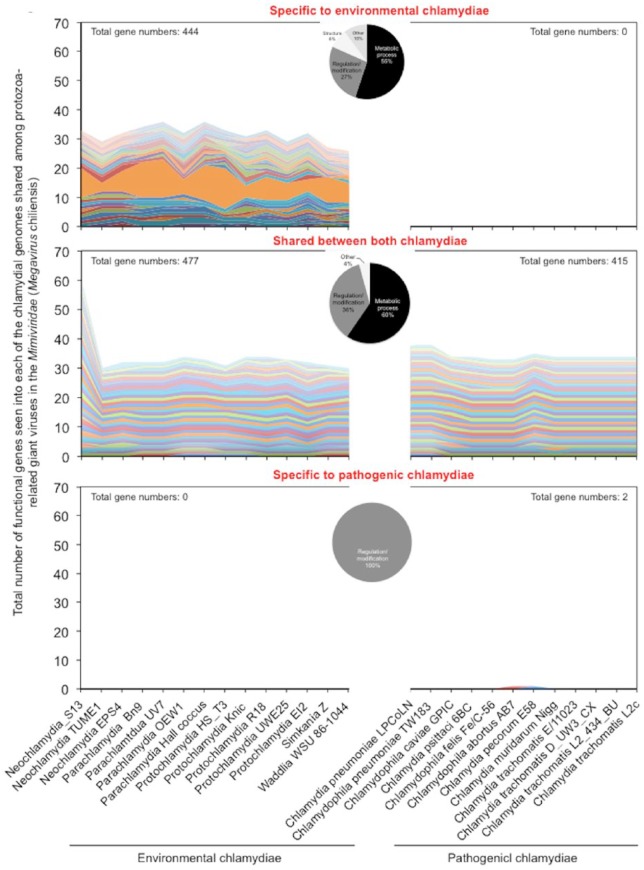
Obligate intracellular chlamydiae diverged into pathogenic and environmental chlamydiae 0.7-1.4 billion years ago. While pathogenic chlamydiae have adapted to a wide range of vertebrates, environmental chlamydiae inhabit unicellular amoebae, the free-living Acanthamoeba. However, how and why this divergence occurred remains unclear. Meanwhile, giant viruses consisting of protozoa-related and protozoa-unrelated viruses have been discovered, with the former group being suggested to have more influenced environmental chlamydiae during their evolution while cohabiting host amoebae. Against this background, we attempted to visualize genes of giant viruses in chlamydial genomes by bioinformatic analysis mainly with comparative genome and phylogenic analysis, seeking genes present in chlamydiae that are specifically shared with protozoa-related giant viruses. As a result, in contrast to protozoa-unrelated giant viruses, the genes of protozoa-related giant viruses were significantly shared in both the chlamydia genomes depending on the giant virus type. In particular, the prevalence of Mimiviridae genes among the protozoa-related giant virus genes in chlamydial genomes was significantly high. Meanwhile, the prevalence of protozoa-related giant virus genes in pathogenic chlamydia genomes was consistently higher than those of environmental chlamydiae; the actual number of sequences similar to giant virus was also significantly predominant compared with those in the environmental chlamydial genomes. Among them, the most prevalent of giant virus was in the case of chlamydiae with Megavirus chiliensis; total of 1338 genes of the chlamydiae were found to be shared with the virus (444 genes specific to environmental chlamydiae, 892 genes shared between both chlamydiae, only two genes in the pathogenic chlamydiae). Phylogenic analysis with most prevalent sets (Megavirus chiliensis and Protochlamydia EI2 or Chlamydia trachomatis L2 434Bu) showed the presence of orthologs between these with several clustered. In addition, Pearson's single regression analysis revealed that almost the prevalence of the genes from the giant viruses in chlamydial genomes was negatively and specifically correlated with the number of chlamydial open reading frames (ORFs). Thus, these results indicated the trace of lateral gene transfer between protozoa-related giant viruses of family Mimiviridae and chlamydiae. This is the first demonstration of a putative linkage between chlamydiae and the giant viruses, providing us with a hint to understand chlamydial evolution.
Keywords: Acanthamoeba; Giant virus; Mimiviridae; environmental chlamydiae; evolution; pathogenic chlamydiae.
Conflict of interest statement
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
Similar articles
-
Related giant viruses in distant locations and different habitats: Acanthamoeba polyphaga moumouvirus represents a third lineage of the Mimiviridae that is close to the megavirus lineage.Genome Biol Evol. 2012;4(12):1324-30. doi: 10.1093/gbe/evs109. Genome Biol Evol. 2012. PMID: 23221609 Free PMC article.
-
Acanthamoeba containing endosymbiotic chlamydia isolated from hospital environments and its potential role in inflammatory exacerbation.BMC Microbiol. 2016 Dec 15;16(1):292. doi: 10.1186/s12866-016-0906-1. BMC Microbiol. 2016. PMID: 27978822 Free PMC article.
-
Complete genome sequence of Courdo11 virus, a member of the family Mimiviridae.Virus Genes. 2014 Apr;48(2):218-23. doi: 10.1007/s11262-013-1016-x. Epub 2013 Dec 1. Virus Genes. 2014. PMID: 24293219
-
Viruses in close associations with free-living amoebae.Parasitol Res. 2015 Nov;114(11):3959-67. doi: 10.1007/s00436-015-4731-5. Epub 2015 Sep 16. Parasitol Res. 2015. PMID: 26374538 Review.
-
Giant mimiviruses escape many canonical criteria of the virus definition.Clin Microbiol Infect. 2019 Feb;25(2):147-154. doi: 10.1016/j.cmi.2018.09.010. Epub 2018 Sep 26. Clin Microbiol Infect. 2019. PMID: 30267933 Review.
Cited by
-
Amoebae as training grounds for microbial pathogens.mBio. 2024 Aug 14;15(8):e0082724. doi: 10.1128/mbio.00827-24. Epub 2024 Jul 8. mBio. 2024. PMID: 38975782 Free PMC article. Review.
-
Giant Viruses-Big Surprises.Viruses. 2019 Apr 30;11(5):404. doi: 10.3390/v11050404. Viruses. 2019. PMID: 31052218 Free PMC article. Review.
References
-
- Horn M. Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol. 2008;62:113–131. - PubMed
-
- Belland RJ, Ouellette SP, Gieffers J, Byrne GI. Chlamydia pneumoniae and atherosclerosis. Cell Microbiol. 2004;6:117–127. - PubMed
-
- Taylor-Brown A, Vaughan L, Greub G, Timms P, Polkinghorne A. Twenty years of research into Chlamydia-like organisms: a revolution in our understanding of the biology and pathogenicity of members of the phylum Chlamydiae. Pathog Dis. 2015;73:1–15. - PubMed
-
- Kebbi-Beghdadi C, Greub G. Importance of amoebae as a tool to isolate amoeba-resisting microorganisms and for their ecology and evolution: the Chlamydia paradigm. Environ Microbiol Rep. 2014;6:309–324. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
