

PIN1 is a new therapeutic target of craniosynostosis
- PMID: 30007339
- PMCID: PMC6216213
- DOI: 10.1093/hmg/ddy252
PIN1 is a new therapeutic target of craniosynostosis
Abstract
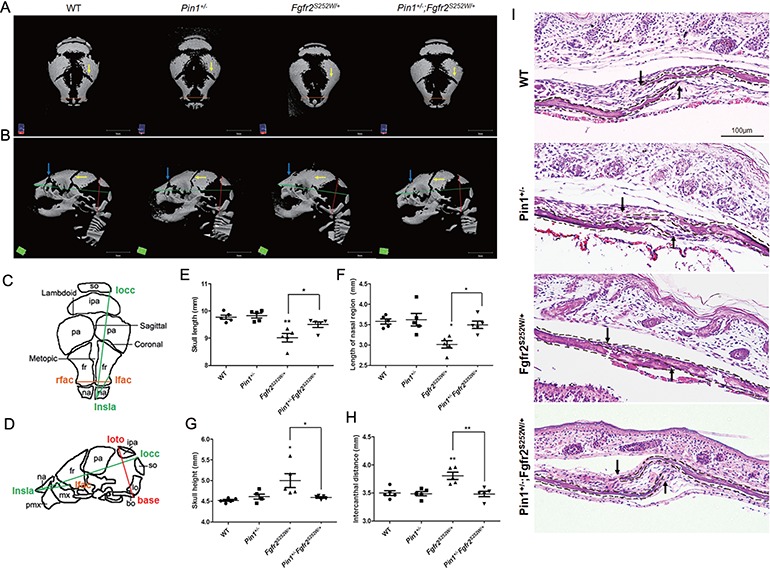
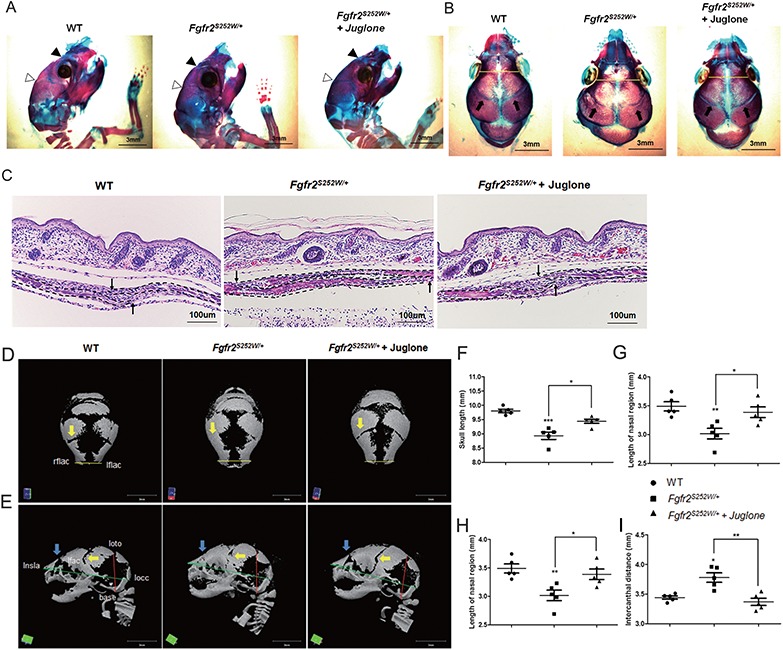
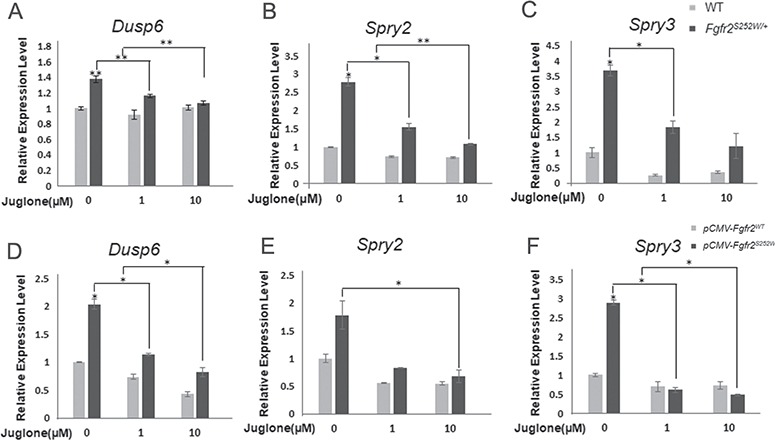
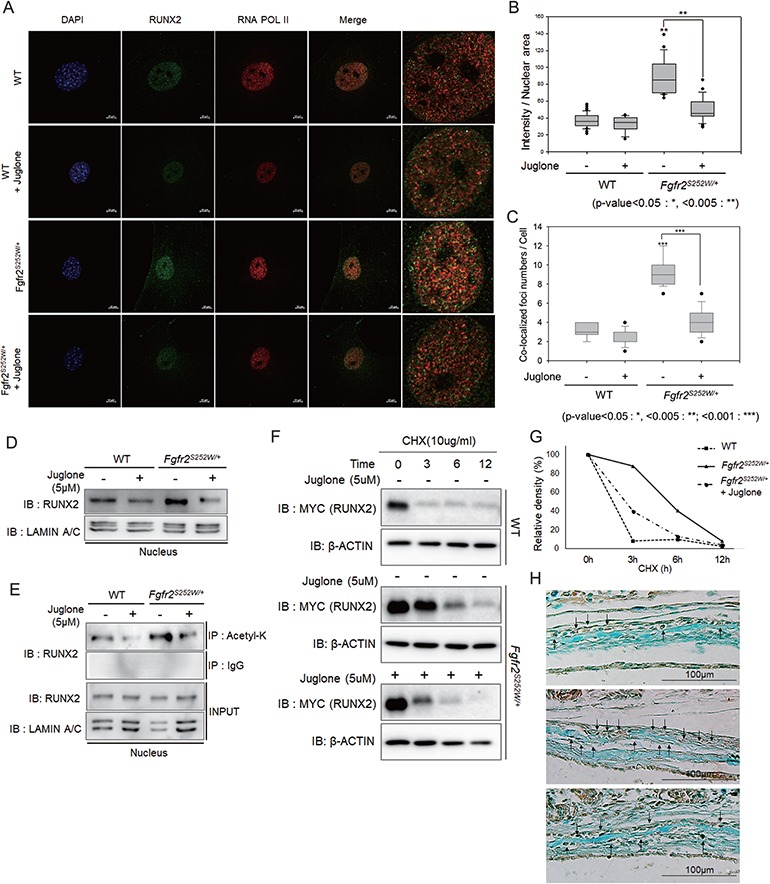
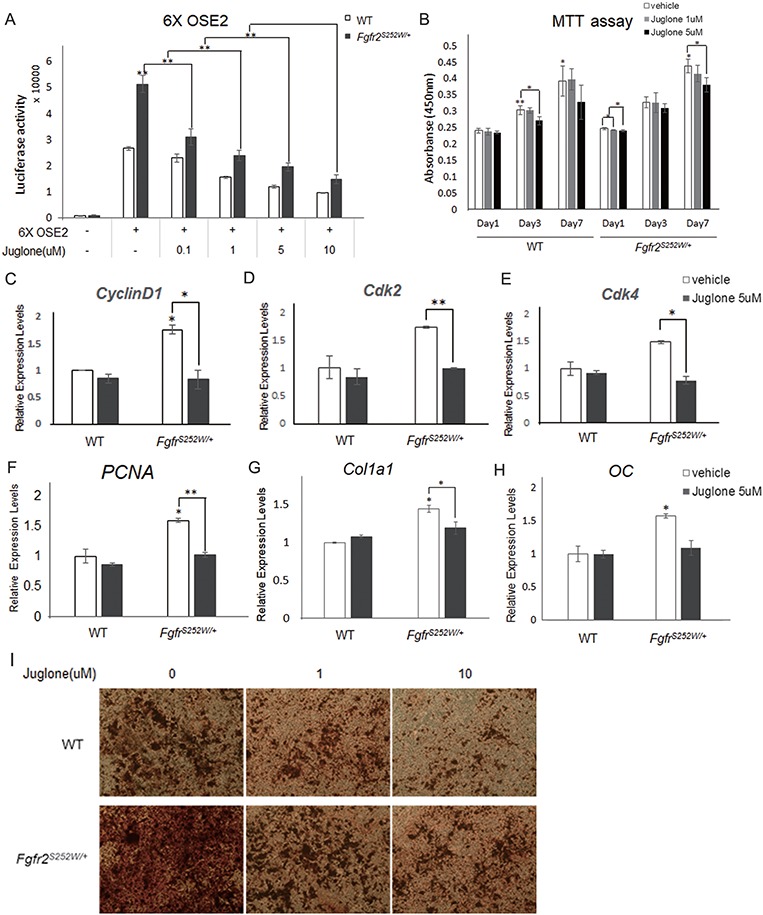
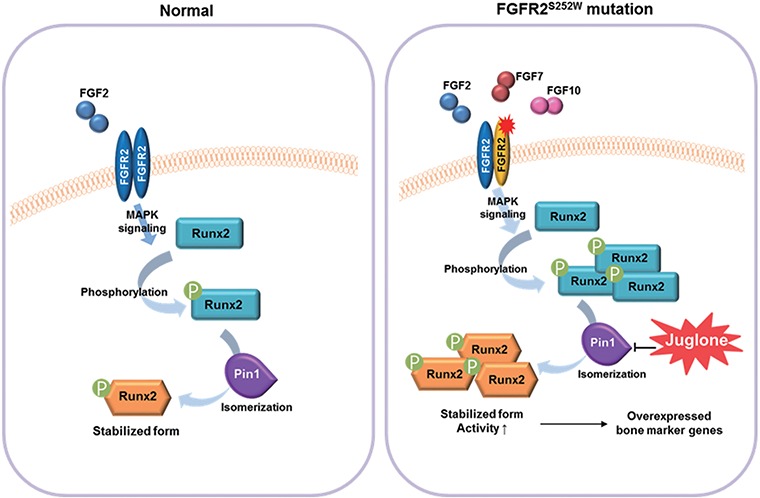
Gain-of-function mutations in fibroblast growth factor receptors (FGFRs) cause congenital skeletal anomalies, including craniosynostosis (CS), which is characterized by the premature closure of craniofacial sutures. Apert syndrome (AS) is one of the severest forms of CS, and the only treatment is surgical expansion of prematurely fused sutures in infants. Previously, we demonstrated that the prolyl isomerase peptidyl-prolyl cis-trans isomerase interacting 1 (PIN1) plays a critical role in mediating FGFR signaling and that Pin1+/- mice exhibit delayed closure of cranial sutures. In this study, using both genetic and pharmacological approaches, we tested whether PIN1 modulation could be used as a therapeutic regimen against AS. In the genetic approach, we crossbred Fgfr2S252W/+, a mouse model of AS, and Pin1+/- mice. Downregulation of Pin1 gene dosage attenuated premature cranial suture closure and other phenotypes of AS in Fgfr2S252W/+ mutant mice. In the pharmacological approach, we intraperitoneally administered juglone, a PIN1 enzyme inhibitor, to pregnant Fgfr2S252W/+ mutant mice and found that this treatment successfully interrupted fetal development of AS phenotypes. Primary cultured osteoblasts from Fgfr2S252W/+ mutant mice expressed high levels of FGFR2 downstream target genes, but this phenotype was attenuated by PIN1 inhibition. Post-translational stabilization and activation of Runt-related transcription factor 2 (RUNX2) in Fgfr2S252W/+ osteoblasts were also attenuated by PIN1 inhibition. Based on these observations, we conclude that PIN1 enzyme activity is important for FGFR2-induced RUNX2 activation and craniofacial suture morphogenesis. Moreover, these findings highlight that juglone or other PIN1 inhibitors represent viable alternatives to surgical intervention for treatment of CS and other hyperostotic diseases.
Figures
Similar articles
-
PIN1 Attenuation Improves Midface Hypoplasia in a Mouse Model of Apert Syndrome.J Dent Res. 2020 Feb;99(2):223-232. doi: 10.1177/0022034519893656. Epub 2019 Dec 23. J Dent Res. 2020. PMID: 31869252
-
Dura in the pathogenesis of syndromic craniosynostosis: fibroblast growth factor receptor 2 mutations in dural cells promote osteogenic proliferation and differentiation of osteoblasts.J Craniofac Surg. 2010 Mar;21(2):462-7. doi: 10.1097/SCS.0b013e3181cfe9a0. J Craniofac Surg. 2010. PMID: 20489451
-
Increased EFG- and PDGFalpha-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis.Hum Mol Genet. 2010 May 1;19(9):1678-89. doi: 10.1093/hmg/ddq045. Epub 2010 Feb 2. Hum Mol Genet. 2010. PMID: 20124286
-
Pin1, the Master Orchestrator of Bone Cell Differentiation.J Cell Physiol. 2017 Sep;232(9):2339-2347. doi: 10.1002/jcp.25442. Epub 2017 Apr 12. J Cell Physiol. 2017. PMID: 27225727 Review.
-
Roles of FGFR2 and twist in human craniosynostosis: insights from genetic mutations in cranial osteoblasts.Front Oral Biol. 2008;12:144-159. doi: 10.1159/000115036. Front Oral Biol. 2008. PMID: 18391499 Review.
Cited by
-
Core biomarkers analysis benefit for diagnosis on human intrahepatic cholestasis of pregnancy.BMC Pregnancy Childbirth. 2024 Aug 10;24(1):525. doi: 10.1186/s12884-024-06730-6. BMC Pregnancy Childbirth. 2024. PMID: 39127651 Free PMC article.
-
Inhibition of miR338 rescues cleidocranial dysplasia in Runx2 mutant mice partially via the Hif1a-Vegfa axis.Exp Mol Med. 2023 Jan;55(1):69-80. doi: 10.1038/s12276-022-00914-w. Epub 2023 Jan 4. Exp Mol Med. 2023. PMID: 36599929 Free PMC article.
-
Excessive osteoclast activation by osteoblast paracrine factor RANKL is a major cause of the abnormal long bone phenotype in Apert syndrome model mice.J Cell Physiol. 2022 Apr;237(4):2155-2168. doi: 10.1002/jcp.30682. Epub 2022 Jan 20. J Cell Physiol. 2022. PMID: 35048384 Free PMC article.
-
Post-Translational Regulations of Transcriptional Activity of RUNX2.Mol Cells. 2020 Feb 29;43(2):160-167. doi: 10.14348/molcells.2019.0247. Mol Cells. 2020. PMID: 31878768 Free PMC article. Review.
-
Insights and future directions of potential genetic therapy for Apert syndrome: A systematic review.Gene Ther. 2021 Nov;28(10-11):620-633. doi: 10.1038/s41434-021-00238-w. Epub 2021 Feb 22. Gene Ther. 2021. PMID: 33619359 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
