

Aberrant Mitochondrial Fission Is Maladaptive in Desmin Mutation-Induced Cardiac Proteotoxicity
- PMID: 29987122
- PMCID: PMC6064863
- DOI: 10.1161/JAHA.118.009289
Aberrant Mitochondrial Fission Is Maladaptive in Desmin Mutation-Induced Cardiac Proteotoxicity
Abstract
Background: Desmin filament proteins interlink the contractile myofibrillar apparatus with mitochondria, nuclei and the sarcolemma. Mutations in the human desmin gene cause cardiac disease, remodeling, and heart failure but the pathophysiological mechanisms remain unknown.
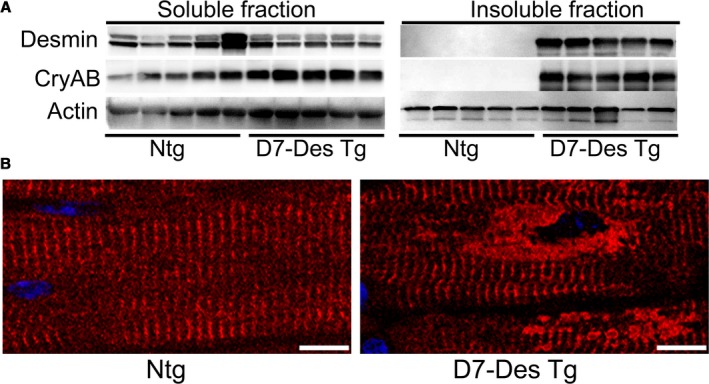
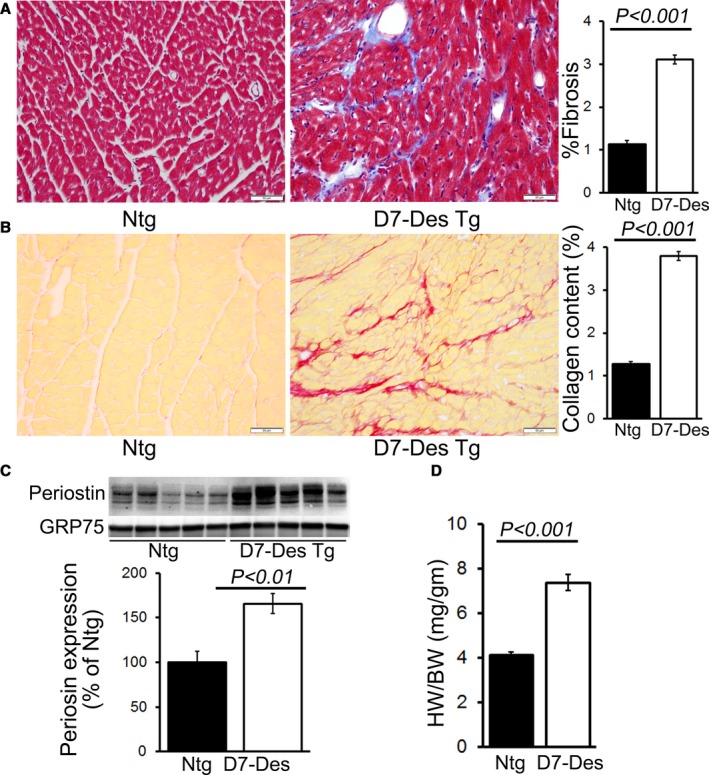
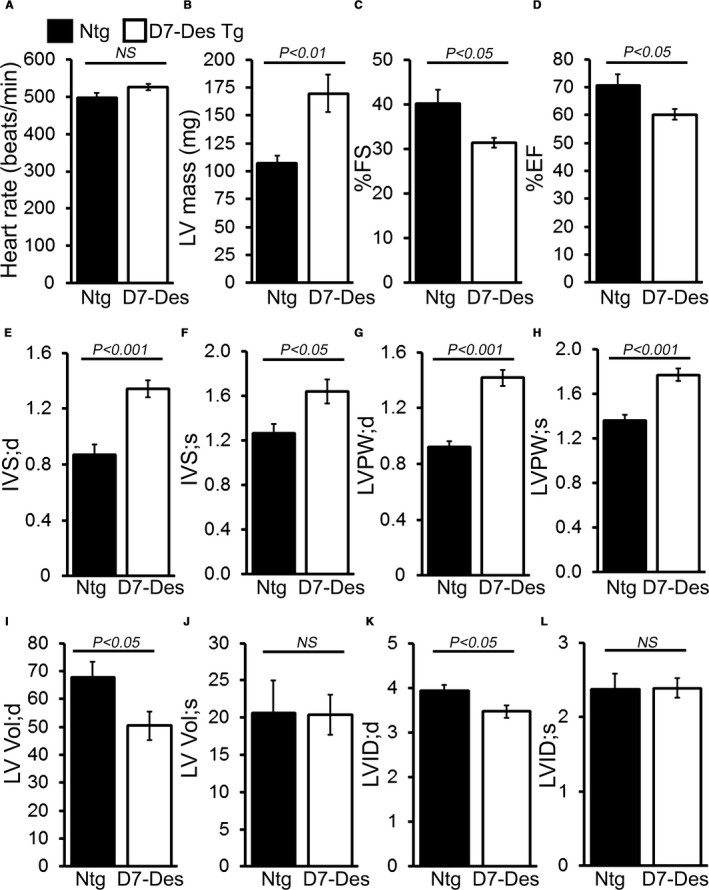
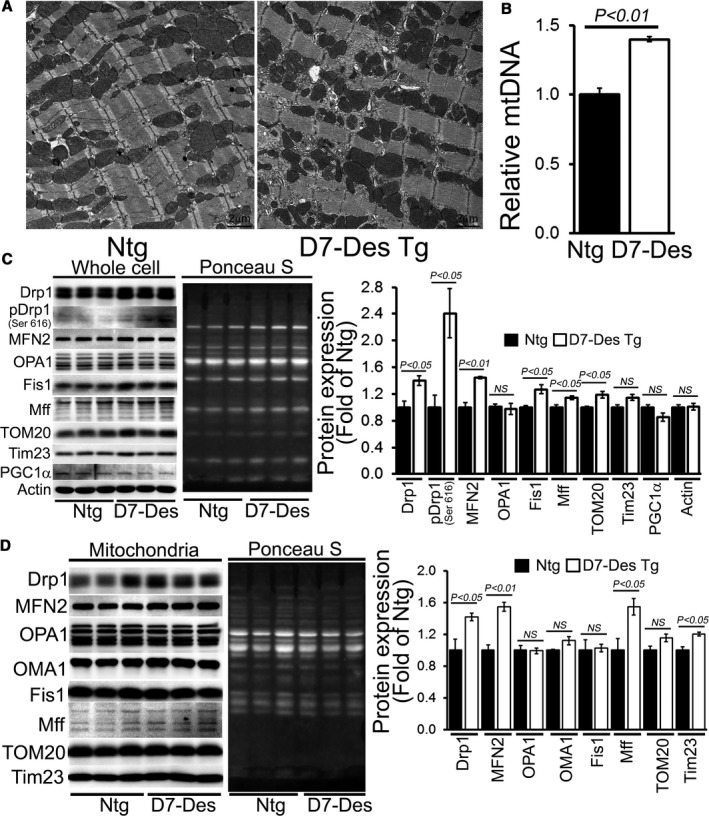
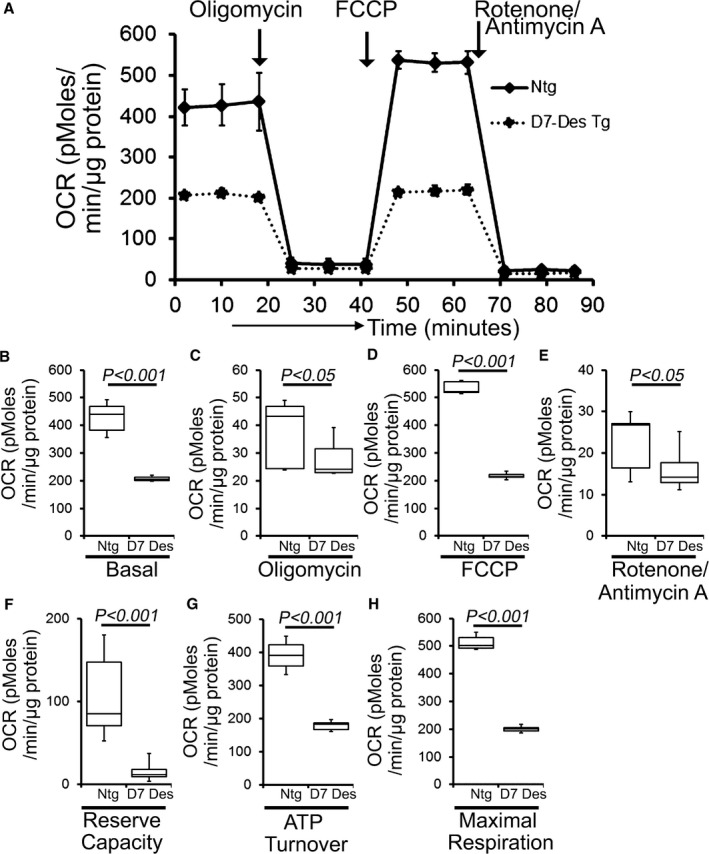
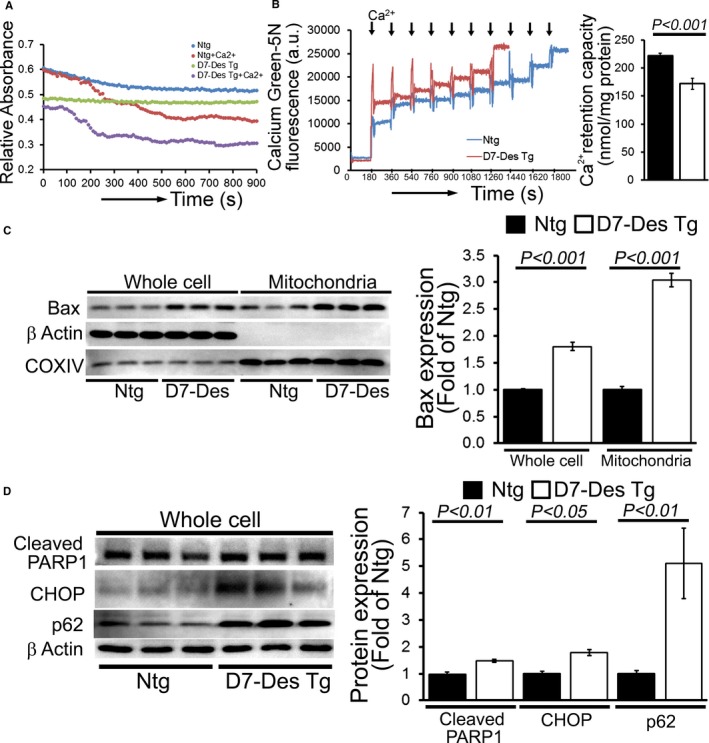
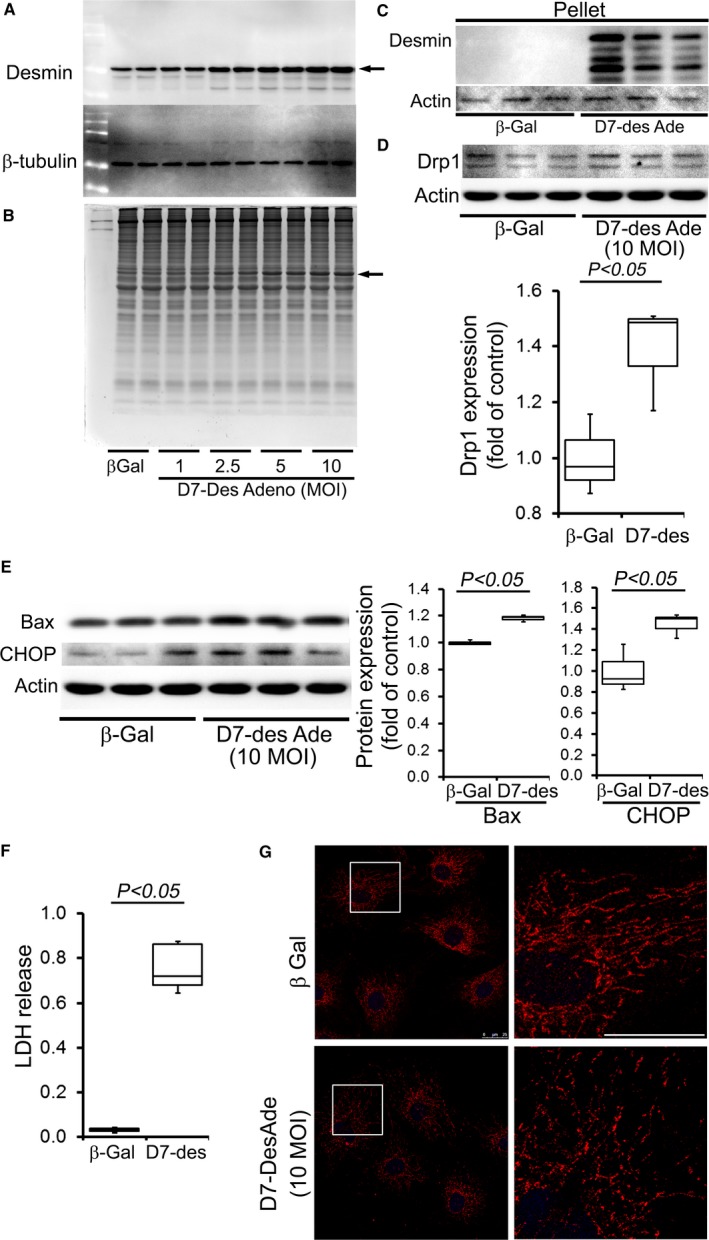
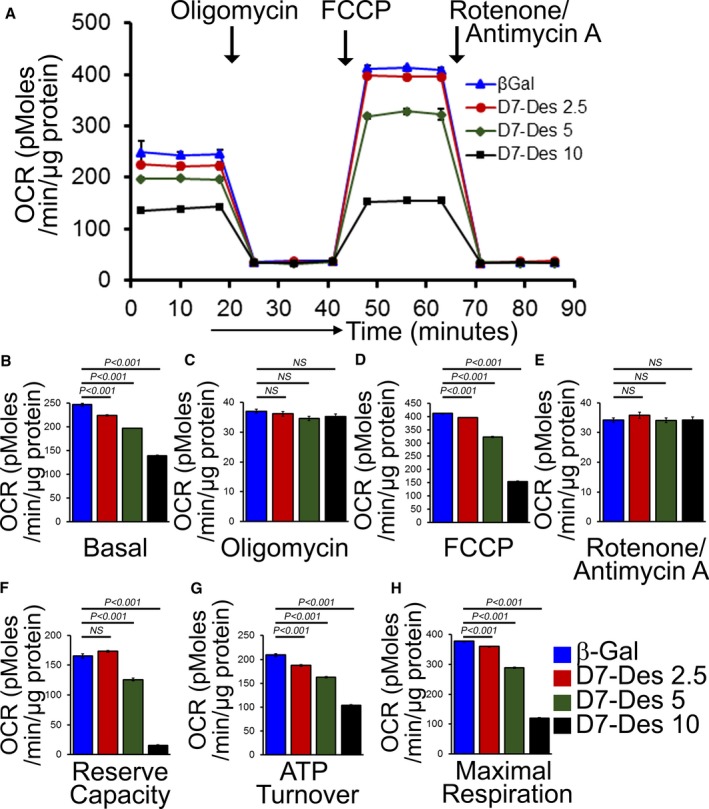
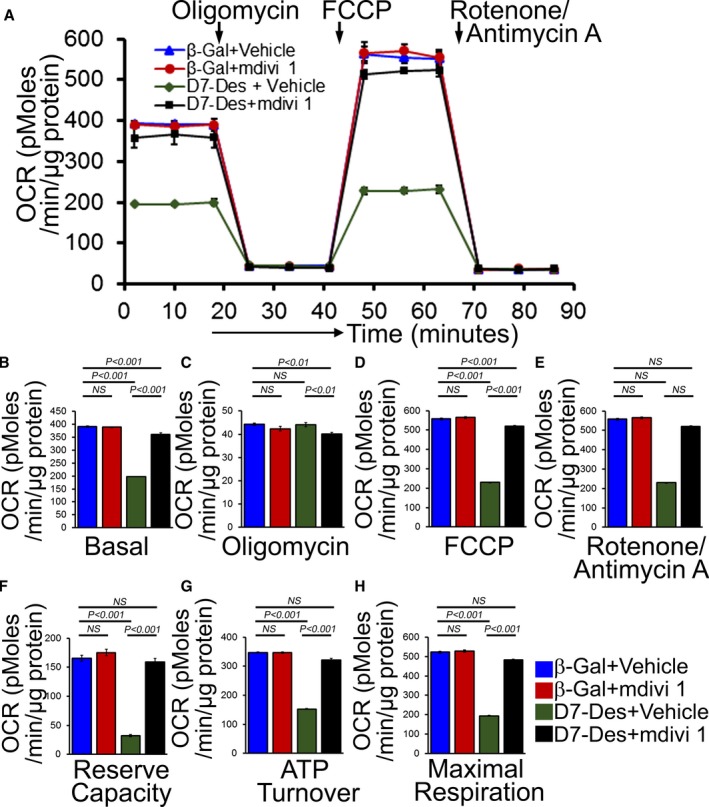
Methods and results: Cardiomyocyte-specific overexpression of mutated desmin (a 7 amino acid deletion R172-E178, D7-Des Tg) causes accumulations of electron-dense aggregates and myofibrillar degeneration associated with cardiac dysfunction. Though extensive studies demonstrated that these altered ultrastructural changes cause impairment of cardiac contractility, the molecular mechanism of cardiomyocyte death remains elusive. In the present study, we report that the D7-Des Tg mouse hearts undergo aberrant mitochondrial fission associated with increased expression of mitochondrial fission regulatory proteins. Mitochondria isolated from D7-Des Tg hearts showed decreased mitochondrial respiration and increased apoptotic cell death. Overexpression of mutant desmin by adenoviral infection in cultured cardiomyocytes led to increased mitochondrial fission, inhibition of mitochondrial respiration, and activation of cellular toxicity. Inhibition of mitochondrial fission by mitochondrial division inhibitor mdivi-1 significantly improved mitochondrial respiration and inhibited cellular toxicity associated with D7-Des overexpression in cardiomyocytes.
Conclusions: Aberrant mitochondrial fission results in mitochondrial respiratory defects and apoptotic cell death in D7-Des Tg hearts. Inhibition of aberrant mitochondrial fission using mitochondrial division inhibitor significantly preserved mitochondrial function and decreased apoptotic cell death. Taken together, our study shows that maladaptive aberrant mitochondrial fission causes desminopathy-associated cellular dysfunction.
Keywords: cardiomyopathy; desminopathy; mitochondrial fission; mitochondrial respiration.
© 2018 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley.
Figures



Similar articles
-
Dysfunctional Mitochondrial Dynamic and Oxidative Phosphorylation Precedes Cardiac Dysfunction in R120G-αB-Crystallin-Induced Desmin-Related Cardiomyopathy.J Am Heart Assoc. 2020 Dec;9(23):e017195. doi: 10.1161/JAHA.120.017195. Epub 2020 Nov 19. J Am Heart Assoc. 2020. PMID: 33208022 Free PMC article.
-
Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia.Circulation. 2018 Apr 10;137(15):1595-1610. doi: 10.1161/CIRCULATIONAHA.117.028719. Epub 2017 Dec 6. Circulation. 2018. PMID: 29212896
-
Biochemical and mechanical dysfunction in a mouse model of desmin-related myopathy.Circ Res. 2009 Apr 24;104(8):1021-8. doi: 10.1161/CIRCRESAHA.108.193516. Epub 2009 Mar 19. Circ Res. 2009. PMID: 19299643 Free PMC article.
-
Pathophysiological mechanisms of cardiomyopathies induced by desmin gene variants located in the C-Terminus of segment 2B.J Cell Physiol. 2024 May;239(5):e31254. doi: 10.1002/jcp.31254. Epub 2024 Mar 19. J Cell Physiol. 2024. PMID: 38501553 Review.
-
Mitochondrial fission/fusion and cardiomyopathy.Curr Opin Genet Dev. 2016 Jun;38:38-44. doi: 10.1016/j.gde.2016.03.001. Epub 2016 Apr 7. Curr Opin Genet Dev. 2016. PMID: 27061490 Free PMC article. Review.
Cited by
-
Desmin gene expression is not ubiquitous in all upper airway myofibers and the pattern differs between healthy and sleep apnea subjects.Eur J Med Res. 2024 Apr 3;29(1):216. doi: 10.1186/s40001-024-01812-9. Eur J Med Res. 2024. PMID: 38566246 Free PMC article.
-
The molecular role of Sigmar1 in regulating mitochondrial function through mitochondrial localization in cardiomyocytes.Mitochondrion. 2022 Jan;62:159-175. doi: 10.1016/j.mito.2021.12.002. Epub 2021 Dec 10. Mitochondrion. 2022. PMID: 34902622 Free PMC article.
-
Mitochondrial dynamics and their potential as a therapeutic target.Mitochondrion. 2019 Nov;49:269-283. doi: 10.1016/j.mito.2019.06.002. Epub 2019 Jun 19. Mitochondrion. 2019. PMID: 31228566 Free PMC article. Review.
-
Desmin Knock-Out Cardiomyopathy: A Heart on the Verge of Metabolic Crisis.Int J Mol Sci. 2022 Oct 10;23(19):12020. doi: 10.3390/ijms231912020. Int J Mol Sci. 2022. PMID: 36233322 Free PMC article.
-
Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration.Sci Rep. 2019 Feb 14;9(1):2002. doi: 10.1038/s41598-018-37862-3. Sci Rep. 2019. PMID: 30765730 Free PMC article.
References
-
- Reipert S, Steinbock F, Fischer I, Bittner RE, Zeold A, Wiche G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp Cell Res. 1999;252:479–491. - PubMed
-
- Bar H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–152. - PubMed
-
- Li Z, Colucci‐Guyon E, Pincon‐Raymond M, Mericskay M, Pournin S, Paulin D, Babinet C. Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol. 1996;175:362–366. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
