

Epigenetic Modifications Associated to Neuroinflammation and Neuropathic Pain After Neural Trauma
- PMID: 29930500
- PMCID: PMC5999732
- DOI: 10.3389/fncel.2018.00158
Epigenetic Modifications Associated to Neuroinflammation and Neuropathic Pain After Neural Trauma
Abstract
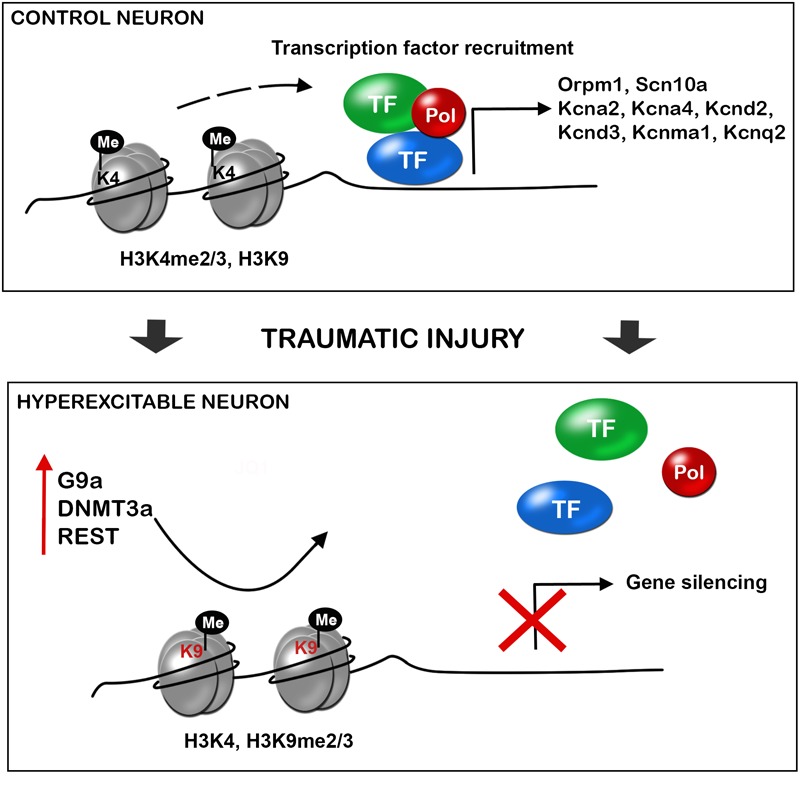
Accumulating evidence suggests that epigenetic alterations lie behind the induction and maintenance of neuropathic pain. Neuropathic pain is usually a chronic condition caused by a lesion, or pathological change, within the nervous system. Neuropathic pain appears frequently after nerve and spinal cord injuries or diseases, producing a debilitation of the patient and a decrease of the quality of life. At the cellular level, neuropathic pain is the result of neuronal plasticity shaped by an increase in the sensitivity and excitability of sensory neurons of the central and peripheral nervous system. One of the mechanisms thought to contribute to hyperexcitability and therefore to the ontogeny of neuropathic pain is the altered expression, trafficking, and functioning of receptors and ion channels expressed by primary sensory neurons. Besides, neuronal and glial cells, such as microglia and astrocytes, together with blood borne macrophages, play a critical role in the induction and maintenance of neuropathic pain by releasing powerful neuromodulators such as pro-inflammatory cytokines and chemokines, which enhance neuronal excitability. Altered gene expression of neuronal receptors, ion channels, and pro-inflammatory cytokines and chemokines, have been associated to epigenetic adaptations of the injured tissue. Within this review, we discuss the involvement of these epigenetic changes, including histone modifications, DNA methylation, non-coding RNAs, and alteration of chromatin modifiers, that have been shown to trigger modification of nociception after neural lesions. In particular, the function on these processes of EZH2, JMJD3, MeCP2, several histone deacetylases (HDACs) and histone acetyl transferases (HATs), G9a, DNMT, REST and diverse non-coding RNAs, are described. Despite the effort on developing new therapies, current treatments have only produced limited relief of this pain in a portion of patients. Thus, the present review aims to contribute to find novel targets for chronic neuropathic pain treatment.
Keywords: epigenetic enzymes; inflammation; neuronal hyperexcitability; neuropathic pain; traumatic injury.
Figures

Similar articles
-
Perspectives in Pain Research 2014: Neuroinflammation and glial cell activation: The cause of transition from acute to chronic pain?Scand J Pain. 2015 Jan 1;6(1):3-6. doi: 10.1016/j.sjpain.2014.10.002. Scand J Pain. 2015. PMID: 29911589
-
Epigenetic Mechanisms of Neural Plasticity in Chronic Neuropathic Pain.ACS Chem Neurosci. 2022 Feb 16;13(4):432-441. doi: 10.1021/acschemneuro.1c00841. Epub 2022 Feb 2. ACS Chem Neurosci. 2022. PMID: 35107991 Review.
-
Elucidation of pathophysiology and treatment of neuropathic pain.Cent Nerv Syst Agents Med Chem. 2012 Dec;12(4):304-14. doi: 10.2174/187152412803760645. Cent Nerv Syst Agents Med Chem. 2012. PMID: 23033930 Review.
-
EZH2 regulates spinal neuroinflammation in rats with neuropathic pain.Neuroscience. 2017 May 4;349:106-117. doi: 10.1016/j.neuroscience.2017.02.041. Epub 2017 Feb 28. Neuroscience. 2017. PMID: 28257897 Free PMC article.
-
Recent advances in understanding neuropathic pain: glia, sex differences, and epigenetics.F1000Res. 2016 Nov 22;5:2743. doi: 10.12688/f1000research.9621.1. eCollection 2016. F1000Res. 2016. PMID: 28105313 Free PMC article. Review.
Cited by
-
CBP Expression Contributes to Neuropathic Pain via CREB and MeCP2 Regulation in the Spared Nerve Injury Rat Model.Medicina (Kaunas). 2024 Jun 17;60(6):989. doi: 10.3390/medicina60060989. Medicina (Kaunas). 2024. PMID: 38929606 Free PMC article.
-
Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins.Biology (Basel). 2023 Jun 26;12(7):914. doi: 10.3390/biology12070914. Biology (Basel). 2023. PMID: 37508346 Free PMC article.
-
Mesenchymal stem cell-derived exosomes: a possible therapeutic strategy for repairing heart injuries.Front Cell Dev Biol. 2023 Jun 30;11:1093113. doi: 10.3389/fcell.2023.1093113. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37457298 Free PMC article. Review.
-
Global research trends on epigenetics and neuropathic pain: A bibliometric analysis.Front Mol Neurosci. 2023 Apr 19;16:1145393. doi: 10.3389/fnmol.2023.1145393. eCollection 2023. Front Mol Neurosci. 2023. PMID: 37152435 Free PMC article. Review.
-
Oxaliplatin-Induced Neuropathy: Genetic and Epigenetic Profile to Better Understand How to Ameliorate This Side Effect.Front Mol Biosci. 2021 May 7;8:643824. doi: 10.3389/fmolb.2021.643824. eCollection 2021. Front Mol Biosci. 2021. PMID: 34026827 Free PMC article. Review.
References
-
- Aldrich B. T., Frakes E. P., Kasuya J., Hammond D. L., Kitamoto T. (2009). Changes in expression of sensory organ-specific microRNAs in rat dorsal root ganglia in association with mechanical hypersensitivity induced by spinal nerve ligation. Neuroscience 164 711–723. 10.1016/j.neuroscience.2009.08.033 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources