

IRE1α Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases
- PMID: 29928282
- PMCID: PMC5997832
- DOI: 10.3389/fimmu.2018.01289
IRE1α Implications in Endoplasmic Reticulum Stress-Mediated Development and Pathogenesis of Autoimmune Diseases
Abstract
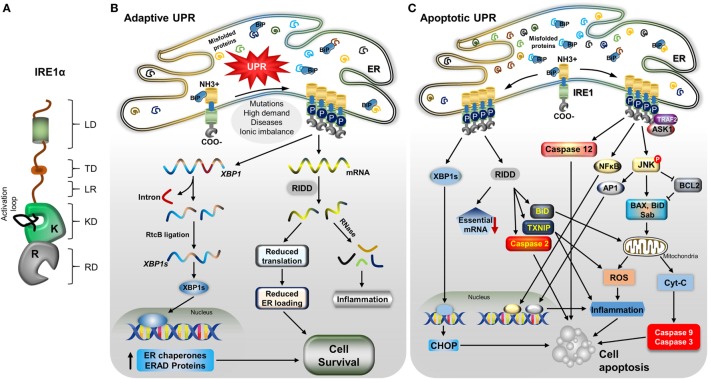
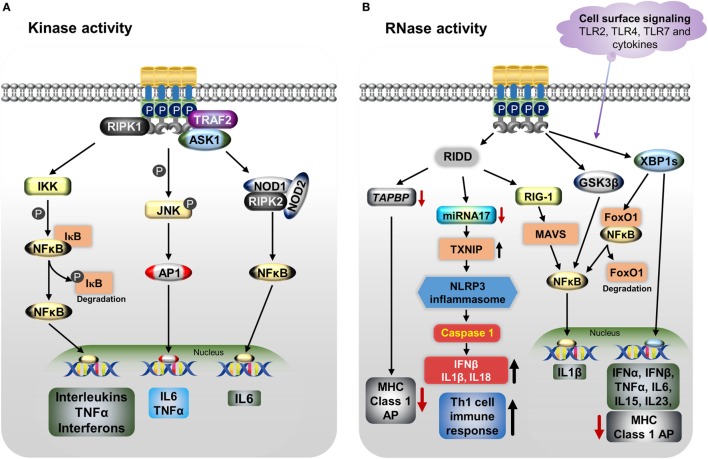
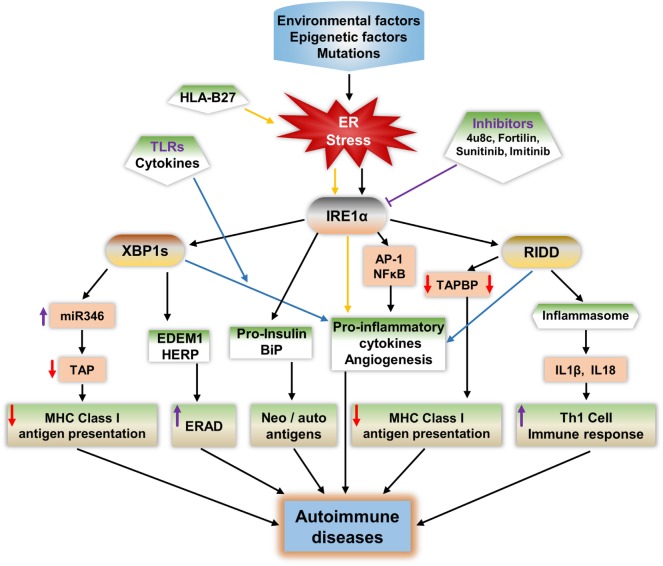
Inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α) is the most prominent and evolutionarily conserved endoplasmic reticulum (ER) membrane protein. This transduces the signal of misfolded protein accumulation in the ER, named as ER stress, to the nucleus as "unfolded protein response (UPR)." The ER stress-mediated IRE1α signaling pathway arbitrates the yin and yang of cell life. IRE1α has been implicated in several physiological as well as pathological conditions, including immune disorders. Autoimmune diseases are caused by abnormal immune responses that develop due to genetic mutations and several environmental factors, including infections and chemicals. These factors dysregulate the cell immune reactions, such as cytokine secretion, antigen presentation, and autoantigen generation. However, the mechanisms involved, in which these factors induce the onset of autoimmune diseases, are remaining unknown. Considering that these environmental factors also induce the UPR, which is expected to have significant role in secretory cells and immune cells. The role of the major UPR molecule, IRE1α, in causing immune responses is well identified, but its role in inducing autoimmunity and the pathogenesis of autoimmune diseases has not been clearly elucidated. Hence, a better understanding of the role of IRE1α and its regulatory mechanisms in causing autoimmune diseases could help to identify and develop the appropriate therapeutic strategies. In this review, we mainly center the discussion on the molecular mechanisms of IRE1α in the pathophysiology of autoimmune diseases.
Keywords: IRE1α; autoimmune diseases; cytokines; inflammation; regulated IRE1α-dependent decay; unfolded protein response signaling pathways.
Figures
Similar articles
-
IRE1α dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development.Cell Signal. 2013 Nov;25(11):2136-46. doi: 10.1016/j.cellsig.2013.06.011. Epub 2013 Jun 29. Cell Signal. 2013. PMID: 23816533
-
Mechanism of the induction of endoplasmic reticulum stress by the anti-cancer agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT): Activation of PERK/eIF2α, IRE1α, ATF6 and calmodulin kinase.Biochem Pharmacol. 2016 Jun 1;109:27-47. doi: 10.1016/j.bcp.2016.04.001. Epub 2016 Apr 6. Biochem Pharmacol. 2016. PMID: 27059255
-
Fortilin binds IRE1α and prevents ER stress from signaling apoptotic cell death.Nat Commun. 2017 May 26;8(1):18. doi: 10.1038/s41467-017-00029-1. Nat Commun. 2017. PMID: 28550308 Free PMC article.
-
Emerging roles for the ER stress sensor IRE1α in metabolic regulation and disease.J Biol Chem. 2019 Dec 6;294(49):18726-18741. doi: 10.1074/jbc.REV119.007036. Epub 2019 Oct 30. J Biol Chem. 2019. PMID: 31666338 Free PMC article. Review.
-
The multiple roles of the unfolded protein response regulator IRE1α in cancer.Mol Carcinog. 2019 Sep;58(9):1623-1630. doi: 10.1002/mc.23031. Epub 2019 Apr 30. Mol Carcinog. 2019. PMID: 31041814 Free PMC article. Review.
Cited by
-
Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development.Pharmaceutics. 2023 Feb 17;15(2):685. doi: 10.3390/pharmaceutics15020685. Pharmaceutics. 2023. PMID: 36840007 Free PMC article. Review.
-
Endoplasmic reticulum stress in innate immune cells - a significant contribution to non-alcoholic fatty liver disease.Front Immunol. 2022 Jul 22;13:951406. doi: 10.3389/fimmu.2022.951406. eCollection 2022. Front Immunol. 2022. PMID: 35958574 Free PMC article. Review.
-
Human and Murine Toll-like Receptor-Driven Disease in Systemic Lupus Erythematosus.Int J Mol Sci. 2024 May 14;25(10):5351. doi: 10.3390/ijms25105351. Int J Mol Sci. 2024. PMID: 38791389 Free PMC article. Review.
-
Protein interaction networks characterizing the A549 cells Klotho transfected are associated with activated pro-apoptotic Bim and suppressed Wnt/β-catenin signaling pathway.Sci Rep. 2024 Jan 25;14(1):2130. doi: 10.1038/s41598-024-52616-0. Sci Rep. 2024. PMID: 38267588 Free PMC article.
-
Asthmatic lung fibroblasts promote type 2 immune responses via endoplasmic reticulum stress response dependent thymic stromal lymphopoietin secretion.Front Physiol. 2023 Jan 25;14:1064822. doi: 10.3389/fphys.2023.1064822. eCollection 2023. Front Physiol. 2023. PMID: 36760534 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical