

Epigenetic maintenance of topological domains in the highly rearranged gibbon genome
- PMID: 29914971
- PMCID: PMC6028127
- DOI: 10.1101/gr.233874.117
Epigenetic maintenance of topological domains in the highly rearranged gibbon genome
Abstract
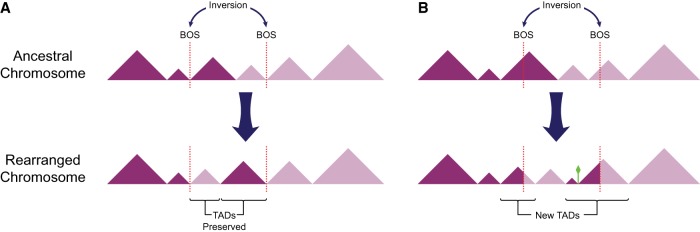
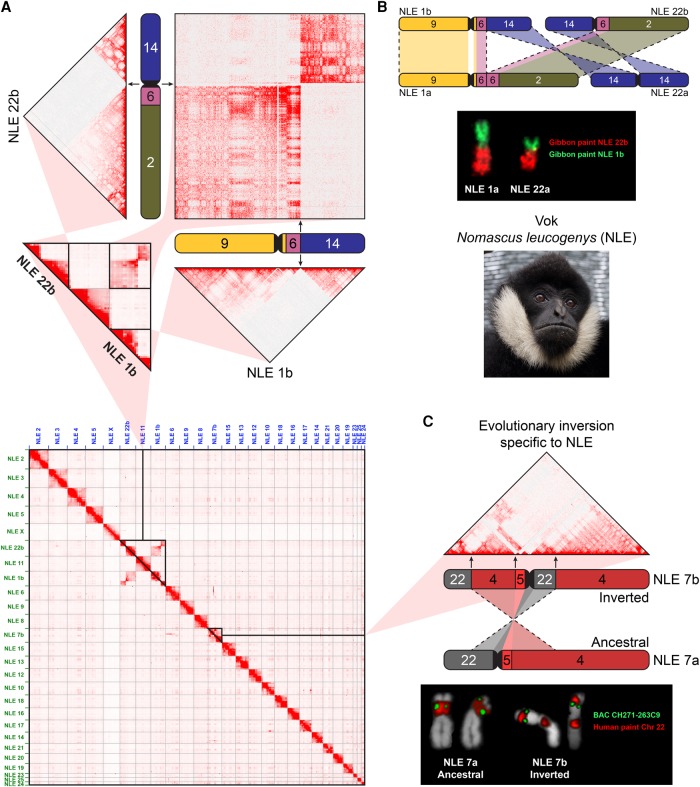
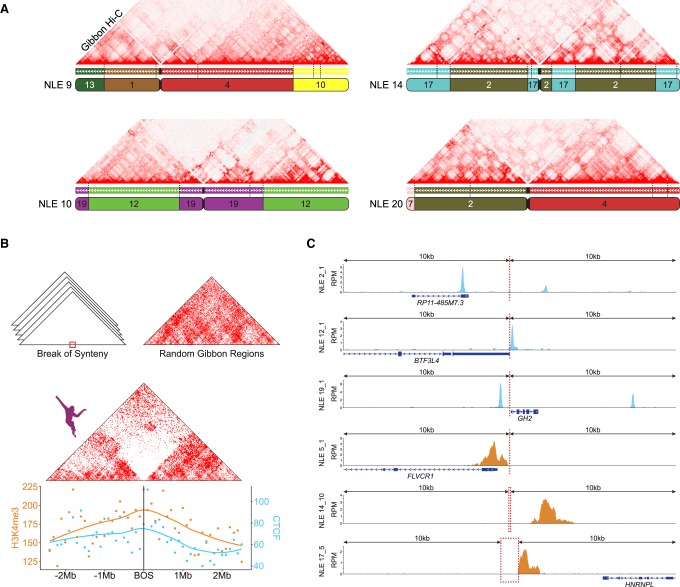
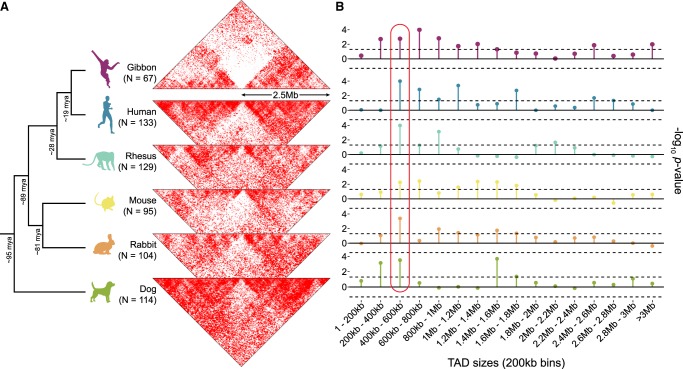
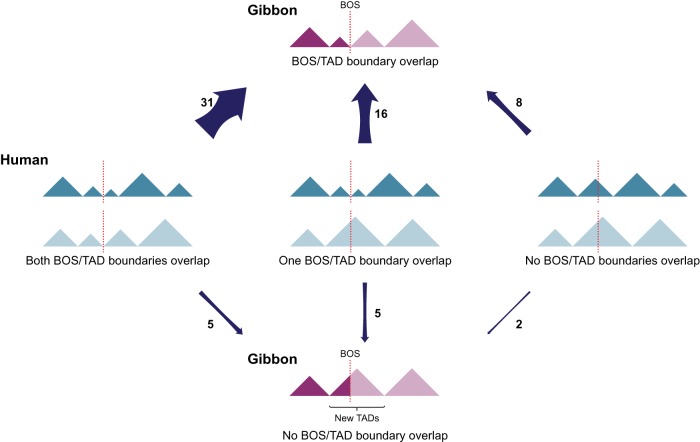
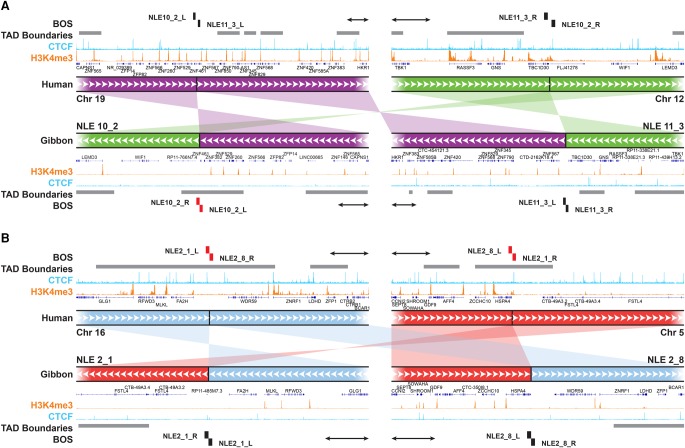
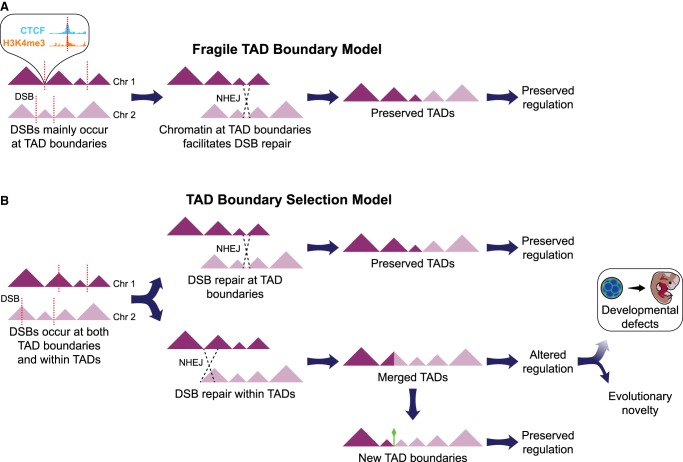
The relationship between evolutionary genome remodeling and the three-dimensional structure of the genome remain largely unexplored. Here, we use the heavily rearranged gibbon genome to examine how evolutionary chromosomal rearrangements impact genome-wide chromatin interactions, topologically associating domains (TADs), and their epigenetic landscape. We use high-resolution maps of gibbon-human breaks of synteny (BOS), apply Hi-C in gibbon, measure an array of epigenetic features, and perform cross-species comparisons. We find that gibbon rearrangements occur at TAD boundaries, independent of the parameters used to identify TADs. This overlap is supported by a remarkable genetic and epigenetic similarity between BOS and TAD boundaries, namely presence of CpG islands and SINE elements, and enrichment in CTCF and H3K4me3 binding. Cross-species comparisons reveal that regions orthologous to BOS also correspond with boundaries of large (400-600 kb) TADs in human and other mammalian species. The colocalization of rearrangement breakpoints and TAD boundaries may be due to higher chromatin fragility at these locations and/or increased selective pressure against rearrangements that disrupt TAD integrity. We also examine the small portion of BOS that did not overlap with TAD boundaries and gave rise to novel TADs in the gibbon genome. We postulate that these new TADs generally lack deleterious consequences. Last, we show that limited epigenetic homogenization occurs across breakpoints, irrespective of their time of occurrence in the gibbon lineage. Overall, our findings demonstrate remarkable conservation of chromatin interactions and epigenetic landscape in gibbons, in spite of extensive genomic shuffling.
© 2018 Lazar et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

Similar articles
-
Genomic rearrangements and evolutionary changes in 3D chromatin topologies in the cotton tribe (Gossypieae).BMC Biol. 2023 Mar 20;21(1):56. doi: 10.1186/s12915-023-01560-y. BMC Biol. 2023. PMID: 36941615 Free PMC article.
-
Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability.Am J Hum Genet. 2021 Feb 4;108(2):269-283. doi: 10.1016/j.ajhg.2021.01.001. Am J Hum Genet. 2021. PMID: 33545030 Free PMC article.
-
TAD evolutionary and functional characterization reveals diversity in mammalian TAD boundary properties and function.Nat Commun. 2023 Dec 7;14(1):8111. doi: 10.1038/s41467-023-43841-8. Nat Commun. 2023. PMID: 38062027 Free PMC article.
-
The connection between BRG1, CTCF and topoisomerases at TAD boundaries.Nucleus. 2017 Mar 4;8(2):150-155. doi: 10.1080/19491034.2016.1276145. Epub 2017 Jan 6. Nucleus. 2017. PMID: 28060558 Free PMC article. Review.
-
Alterations in Three-Dimensional Organization of the Cancer Genome and Epigenome.Cold Spring Harb Symp Quant Biol. 2016;81:41-51. doi: 10.1101/sqb.2016.81.031013. Epub 2017 Apr 19. Cold Spring Harb Symp Quant Biol. 2016. PMID: 28424341 Review.
Cited by
-
Chromatin domain alterations linked to 3D genome organization in a large cohort of schizophrenia and bipolar disorder brains.Nat Neurosci. 2022 Apr;25(4):474-483. doi: 10.1038/s41593-022-01032-6. Epub 2022 Mar 24. Nat Neurosci. 2022. PMID: 35332326 Free PMC article.
-
Epigenomic translocation of H3K4me3 broad domains over oncogenes following hijacking of super-enhancers.Genome Res. 2022 Jul;32(7):1343-1354. doi: 10.1101/gr.276042.121. Epub 2021 Dec 21. Genome Res. 2022. PMID: 34933939 Free PMC article.
-
The Genome of Blue-Capped Cordon-Bleu Uncovers Hidden Diversity of LTR Retrotransposons in Zebra Finch.Genes (Basel). 2019 Apr 13;10(4):301. doi: 10.3390/genes10040301. Genes (Basel). 2019. PMID: 31013951 Free PMC article.
-
Chromatin spatial organization of wild type and mutant peanuts reveals high-resolution genomic architecture and interaction alterations.Genome Biol. 2021 Nov 16;22(1):315. doi: 10.1186/s13059-021-02520-x. Genome Biol. 2021. PMID: 34784945 Free PMC article.
-
TAD fusion score: discovery and ranking the contribution of deletions to genome structure.Genome Biol. 2019 Mar 21;20(1):60. doi: 10.1186/s13059-019-1666-7. Genome Biol. 2019. PMID: 30898144 Free PMC article.
References
-
- Berthelot C, Muffato M, Abecassis J, Roest Crollius H. 2015. The 3D organization of chromatin explains evolutionary fragile genomic regions. Cell Rep 10: 1913–1924. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases