

Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction
- PMID: 29754270
- PMCID: PMC5973959
- DOI: 10.1007/s00439-018-1880-5
Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction
Abstract
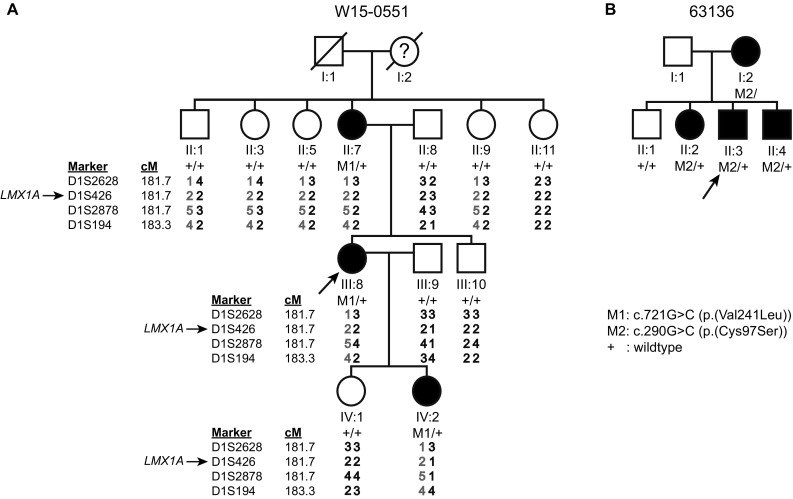
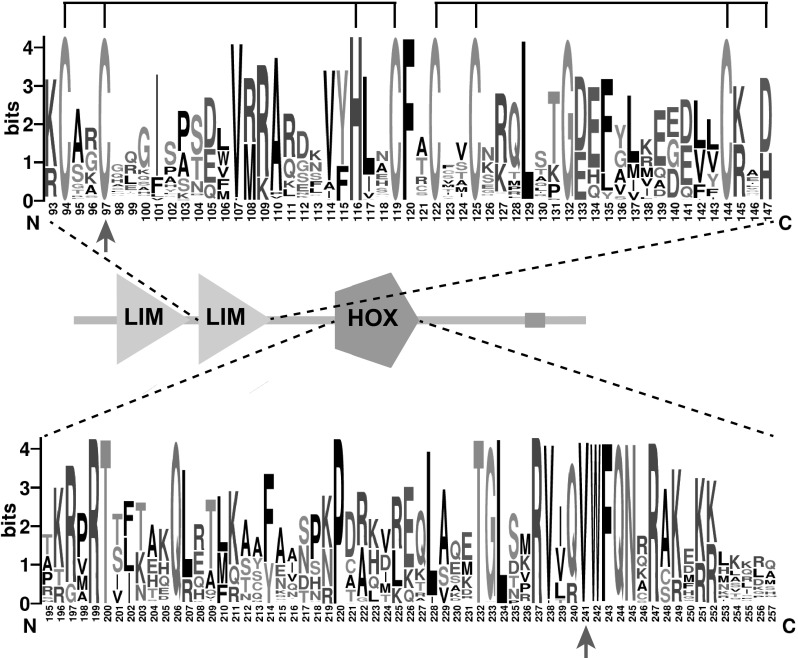
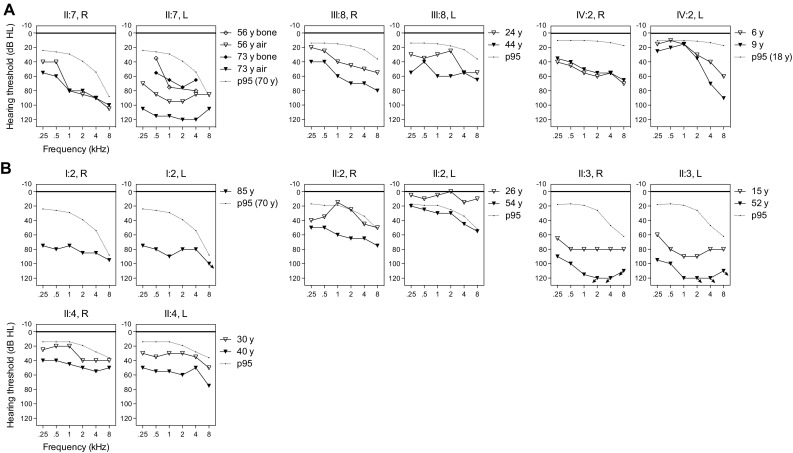
Unraveling the causes and pathomechanisms of progressive disorders is essential for the development of therapeutic strategies. Here, we identified heterozygous pathogenic missense variants of LMX1A in two families of Dutch origin with progressive nonsyndromic hearing impairment (HI), using whole exome sequencing. One variant, c.721G > C (p.Val241Leu), occurred de novo and is predicted to affect the homeodomain of LMX1A, which is essential for DNA binding. The second variant, c.290G > C (p.Cys97Ser), predicted to affect a zinc-binding residue of the second LIM domain that is involved in protein-protein interactions. Bi-allelic deleterious variants of Lmx1a are associated with a complex phenotype in mice, including deafness and vestibular defects, due to arrest of inner ear development. Although Lmx1a mouse mutants demonstrate neurological, skeletal, pigmentation and reproductive system abnormalities, no syndromic features were present in the participating subjects of either family. LMX1A has previously been suggested as a candidate gene for intellectual disability, but our data do not support this, as affected subjects displayed normal cognition. Large variability was observed in the age of onset (a)symmetry, severity and progression rate of HI. About half of the affected individuals displayed vestibular dysfunction and experienced symptoms thereof. The late-onset progressive phenotype and the absence of cochleovestibular malformations on computed tomography scans indicate that heterozygous defects of LMX1A do not result in severe developmental abnormalities in humans. We propose that a single LMX1A wild-type copy is sufficient for normal development but insufficient for maintenance of cochleovestibular function. Alternatively, minor cochleovestibular developmental abnormalities could eventually lead to the progressive phenotype seen in the families.
Conflict of interest statement
The authors have no conflict of interest to declare.
Figures
Similar articles
-
A variant in LMX1A causes autosomal recessive severe-to-profound hearing impairment.Hum Genet. 2018 Jul;137(6-7):471-478. doi: 10.1007/s00439-018-1899-7. Epub 2018 Jul 3. Hum Genet. 2018. PMID: 29971487 Free PMC article.
-
Novel Molecular Genetic Etiology of Asymmetric Hearing Loss: Autosomal-Dominant LMX1A Variants.Ear Hear. 2022 Nov-Dec 01;43(6):1698-1707. doi: 10.1097/AUD.0000000000001237. Epub 2022 Jun 17. Ear Hear. 2022. PMID: 35711095
-
Novel genotype-phenotype correlation of functionally characterized LMX1A variants linked to sensorineural hearing loss.Hum Mutat. 2020 Nov;41(11):1877-1883. doi: 10.1002/humu.24095. Epub 2020 Sep 9. Hum Mutat. 2020. PMID: 32840933
-
[From gene to disease; a progressive cochlear-vestibular dysfunction with onset in middle-age (DFNA9)].Ned Tijdschr Geneeskd. 2005 Nov 19;149(47):2619-21. Ned Tijdschr Geneeskd. 2005. PMID: 16355574 Review. Dutch.
-
Expansion of the phenotypic spectrum associated with pathogenic missense variation in DHX16.Am J Med Genet A. 2024 Jan;194(1):53-58. doi: 10.1002/ajmg.a.63392. Epub 2023 Sep 4. Am J Med Genet A. 2024. PMID: 37664979 Review.
Cited by
-
Identification of Novel Candidate Genes and Variants for Hearing Loss and Temporal Bone Anomalies.Genes (Basel). 2021 Apr 13;12(4):566. doi: 10.3390/genes12040566. Genes (Basel). 2021. PMID: 33924653 Free PMC article.
-
Exome variant prioritization in a large cohort of hearing-impaired individuals indicates IKZF2 to be associated with non-syndromic hearing loss and guides future research of unsolved cases.Hum Genet. 2024 Nov;143(11):1379-1399. doi: 10.1007/s00439-024-02706-w. Epub 2024 Oct 16. Hum Genet. 2024. PMID: 39406892 Free PMC article.
-
A variant in LMX1A causes autosomal recessive severe-to-profound hearing impairment.Hum Genet. 2018 Jul;137(6-7):471-478. doi: 10.1007/s00439-018-1899-7. Epub 2018 Jul 3. Hum Genet. 2018. PMID: 29971487 Free PMC article.
-
Interaction with ectopic cochlear crista sensory epithelium disrupts basal cochlear sensory epithelium development in Lmx1a mutant mice.Cell Tissue Res. 2020 Jun;380(3):435-448. doi: 10.1007/s00441-019-03163-y. Epub 2020 Jan 13. Cell Tissue Res. 2020. PMID: 31932950 Free PMC article.
-
Autosomal Dominant Non-Syndromic Hearing Loss (DFNA): A Comprehensive Narrative Review.Biomedicines. 2023 Jun 1;11(6):1616. doi: 10.3390/biomedicines11061616. Biomedicines. 2023. PMID: 37371710 Free PMC article. Review.
References
-
- Bongers EM, de Wijs IJ, Marcelis C, Hoefsloot LH, Knoers NV. Identification of entire LMX1B gene deletions in nail patella syndrome: evidence for haploinsufficiency as the main pathogenic mechanism underlying dominant inheritance in man. Eur J Hum Genet EJHG. 2008;16:1240–1244. doi: 10.1038/ejhg.2008.83. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
