

Time-resolved transcriptome and proteome landscape of human regulatory T cell (Treg) differentiation reveals novel regulators of FOXP3
- PMID: 29730990
- PMCID: PMC5937035
- DOI: 10.1186/s12915-018-0518-3
Time-resolved transcriptome and proteome landscape of human regulatory T cell (Treg) differentiation reveals novel regulators of FOXP3
Abstract
Background: Regulatory T cells (Tregs) expressing the transcription factor FOXP3 are crucial mediators of self-tolerance, preventing autoimmune diseases but possibly hampering tumor rejection. Clinical manipulation of Tregs is of great interest, and first-in-man trials of Treg transfer have achieved promising outcomes. Yet, the mechanisms governing induced Treg (iTreg) differentiation and the regulation of FOXP3 are incompletely understood.
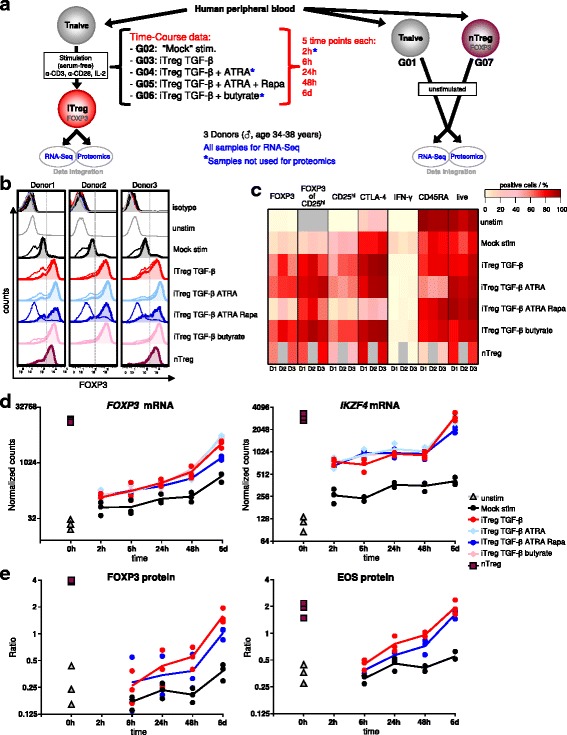
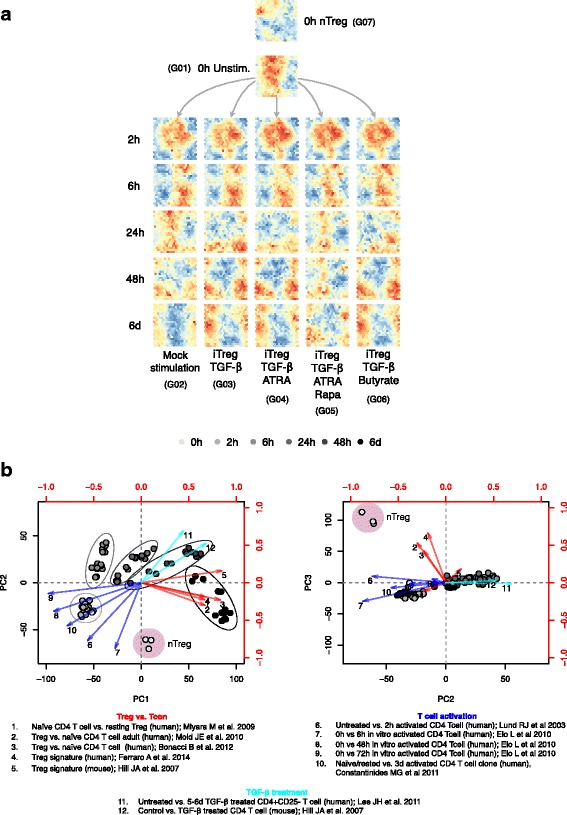
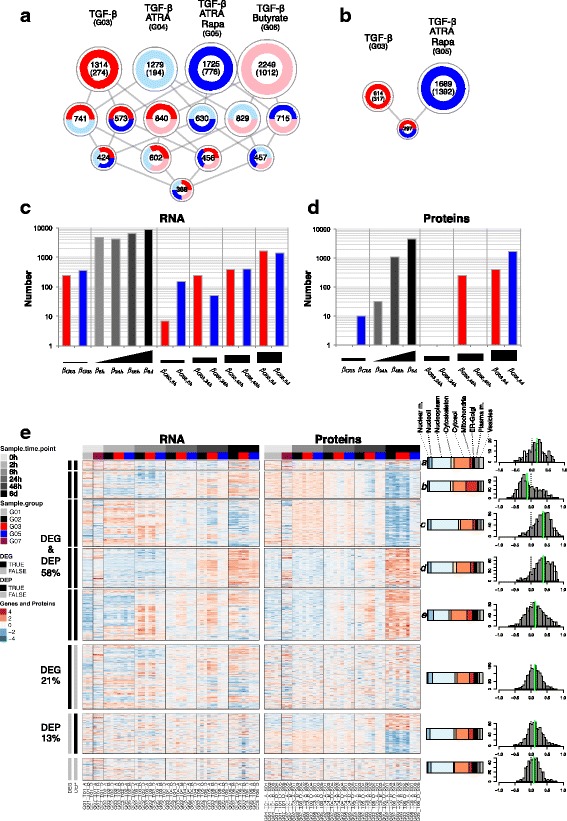
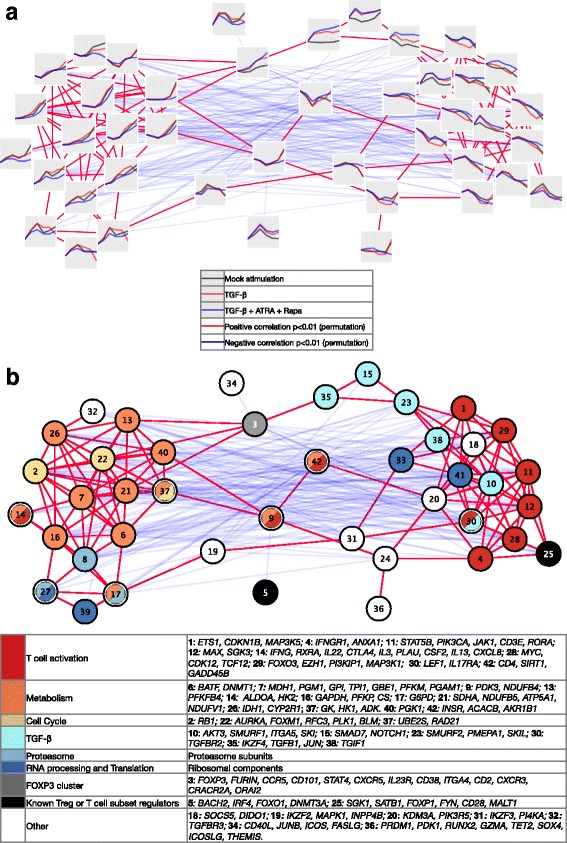
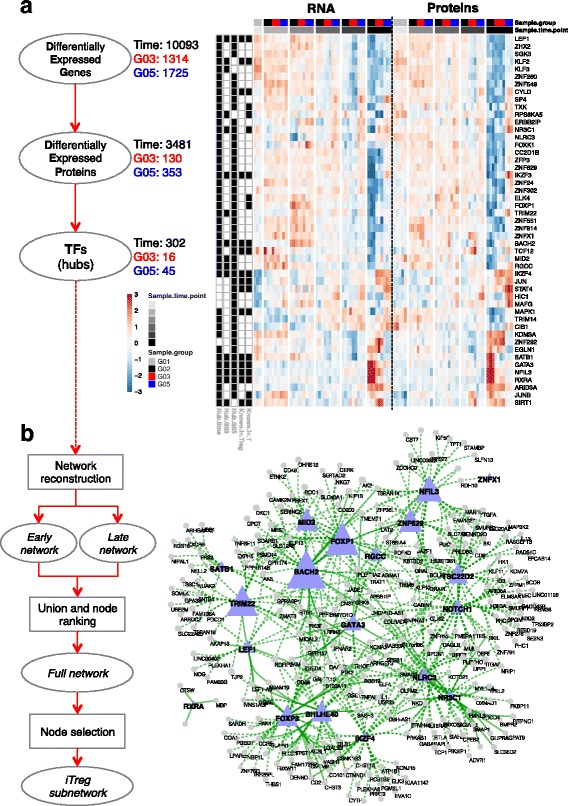
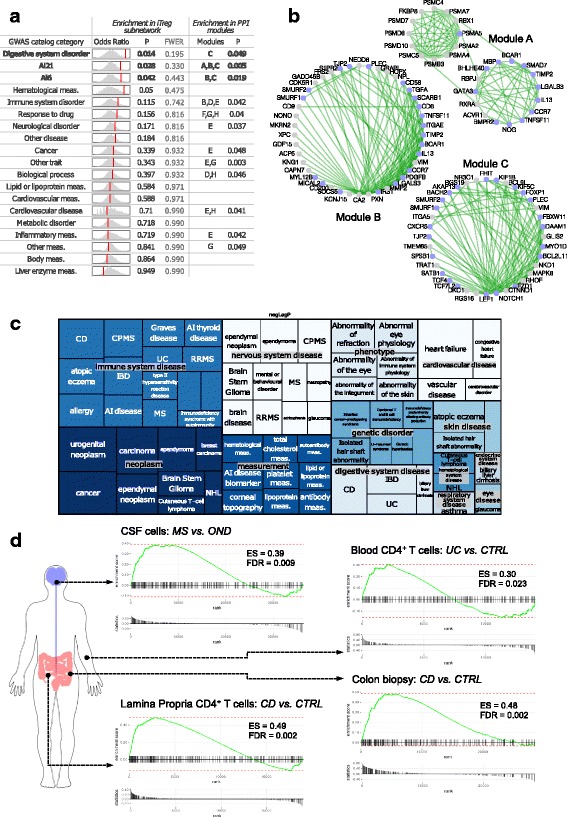
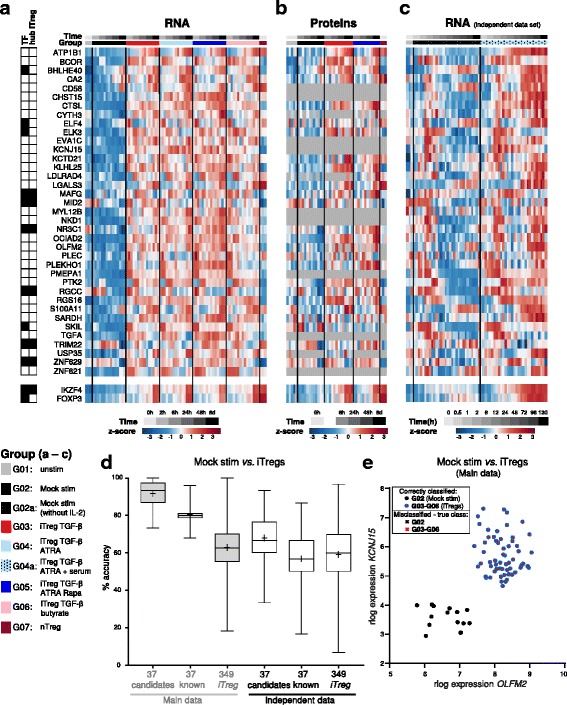
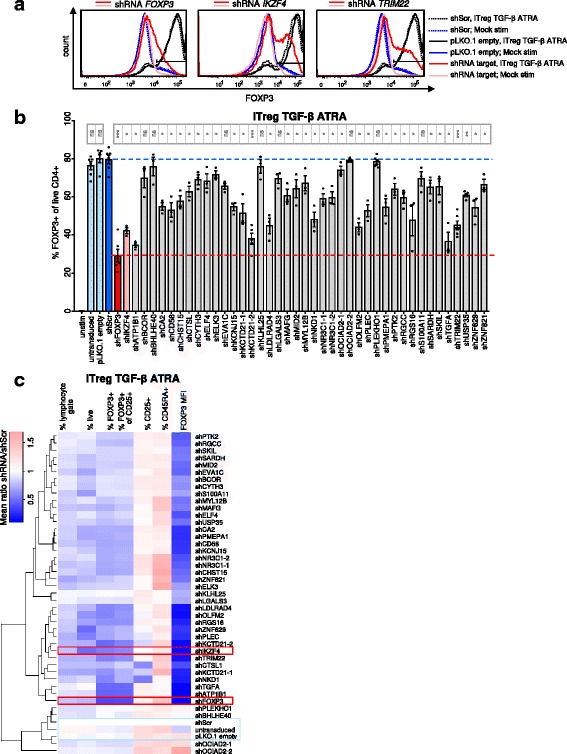
Results: To gain a comprehensive and unbiased molecular understanding of FOXP3 induction, we performed time-series RNA sequencing (RNA-Seq) and proteomics profiling on the same samples during human iTreg differentiation. To enable the broad analysis of universal FOXP3-inducing pathways, we used five differentiation protocols in parallel. Integrative analysis of the transcriptome and proteome confirmed involvement of specific molecular processes, as well as overlap of a novel iTreg subnetwork with known Treg regulators and autoimmunity-associated genes. Importantly, we propose 37 novel molecules putatively involved in iTreg differentiation. Their relevance was validated by a targeted shRNA screen confirming a functional role in FOXP3 induction, discriminant analyses classifying iTregs accordingly, and comparable expression in an independent novel iTreg RNA-Seq dataset.
Conclusion: The data generated by this novel approach facilitates understanding of the molecular mechanisms underlying iTreg generation as well as of the concomitant changes in the transcriptome and proteome. Our results provide a reference map exploitable for future discovery of markers and drug candidates governing control of Tregs, which has important implications for the treatment of cancer, autoimmune, and inflammatory diseases.
Keywords: Data integration; FOXP3; Proteomics; RNA sequencing (RNA-Seq); Regulatory T cells; T cell differentiation; TGF-β; Treg; iTreg.
Conflict of interest statement
Ethics approval and consent to participate
The experimental methods comply with the Declaration of Helsinki and ethical approval was obtained as appropriate and is detailed in the respective Methods sections.
Competing interests
CCG received speaker honoraria and travel expenses for attending meetings from Bayer Health Care, Genzyme, and Novartis Pharma GmbH and her research is funded by the Interdisciplinary Center for Clinical Studies (IZKF). HW receives honoraria for acting as a member of Scientific Advisory Boards and as consultant for Biogen, Evgen, MedDay Pharmaceuticals, Merck Serono, Novartis, Roche Pharma AG, Sanofi-Genzyme, as well as speaker honoraria and travel support from Alexion, Biogen, Cognomed, F. Hoffmann-La Roche Ltd., Gemeinnützige Hertie-Stiftung, Merck Serono, Novartis, Roche Pharma AG, Sanofi-Genzyme, TEVA, and WebMD Global. HW is acting as a paid consultant for Abbvie, Actelion, Biogen, IGES, Novartis, Roche, Sanofi-Genzyme, and the Swiss Multiple Sclerosis Society. HW received research funding from the Else Kröner Fresenius Foundation, Fresenius Foundation, Hertie Foundation, NRW Ministry of Education and Research, IZKF Muenster and RE Children’s Foundation, Biogen GmbH, GlaxoSmithKline GmbH, Roche Pharma AG, and Sanofi-Genzyme.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
All other authors declare no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Epigenetic conversion of conventional T cells into regulatory T cells by CD28 signal deprivation.Proc Natl Acad Sci U S A. 2020 Jun 2;117(22):12258-12268. doi: 10.1073/pnas.1922600117. Epub 2020 May 15. Proc Natl Acad Sci U S A. 2020. PMID: 32414925 Free PMC article.
-
In Vitro Differentiation of Human CD4+FOXP3+ Induced Regulatory T Cells (iTregs) from Naïve CD4+ T Cells Using a TGF-β-containing Protocol.J Vis Exp. 2016 Dec 30;(118):55015. doi: 10.3791/55015. J Vis Exp. 2016. PMID: 28060341 Free PMC article.
-
Critical Role of TGF-β and IL-2 Receptor Signaling in Foxp3 Induction by an Inhibitor of DNA Methylation.Front Immunol. 2018 Feb 2;9:125. doi: 10.3389/fimmu.2018.00125. eCollection 2018. Front Immunol. 2018. PMID: 29456534 Free PMC article.
-
Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes.Immunology. 2020 May;160(1):24-37. doi: 10.1111/imm.13178. Epub 2020 Mar 9. Immunology. 2020. PMID: 32022254 Free PMC article. Review.
-
From stability to dynamics: understanding molecular mechanisms of regulatory T cells through Foxp3 transcriptional dynamics.Clin Exp Immunol. 2019 Jul;197(1):14-23. doi: 10.1111/cei.13194. Epub 2018 Sep 17. Clin Exp Immunol. 2019. PMID: 30076771 Free PMC article. Review.
Cited by
-
Substrate stiffness enhances human regulatory T cell induction and metabolism.Biomaterials. 2023 Jan;292:121928. doi: 10.1016/j.biomaterials.2022.121928. Epub 2022 Nov 26. Biomaterials. 2023. PMID: 36455488 Free PMC article.
-
An elastic-net logistic regression approach to generate classifiers and gene signatures for types of immune cells and T helper cell subsets.BMC Bioinformatics. 2019 Aug 22;20(1):433. doi: 10.1186/s12859-019-2994-z. BMC Bioinformatics. 2019. PMID: 31438843 Free PMC article.
-
GeneCloudOmics: A Data Analytic Cloud Platform for High-Throughput Gene Expression Analysis.Front Bioinform. 2021 Nov 25;1:693836. doi: 10.3389/fbinf.2021.693836. eCollection 2021. Front Bioinform. 2021. PMID: 36303746 Free PMC article.
-
Proteomics reveals unique identities of human TGF-β-induced and thymus-derived CD4+ regulatory T cells.Sci Rep. 2022 Nov 24;12(1):20268. doi: 10.1038/s41598-022-23515-z. Sci Rep. 2022. PMID: 36434024 Free PMC article.
-
The implications and prospect of cuproptosis-related genes and copper transporters in cancer progression.Front Oncol. 2023 Feb 28;13:1117164. doi: 10.3389/fonc.2023.1117164. eCollection 2023. Front Oncol. 2023. PMID: 36925927 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
