

The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes
- PMID: 29728366
- PMCID: PMC5991525
- DOI: 10.1101/gr.228171.117
The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes
Abstract
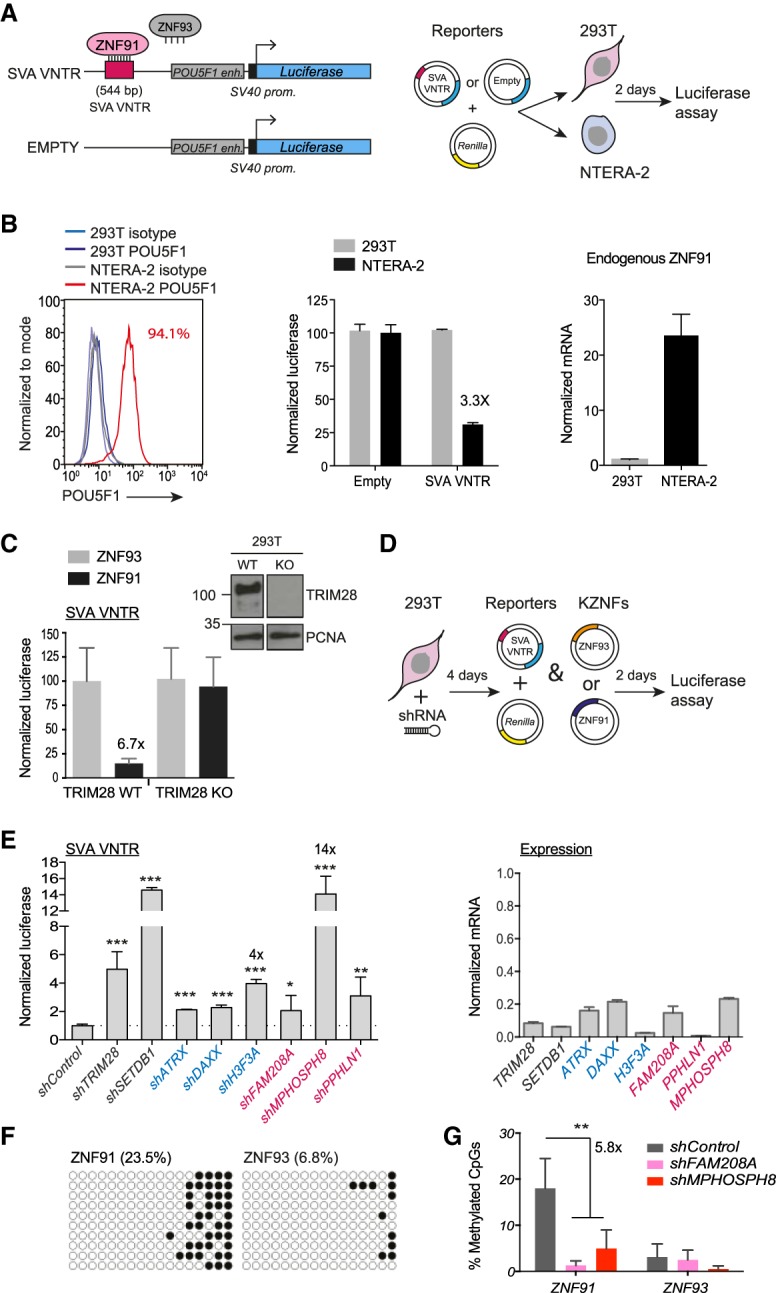
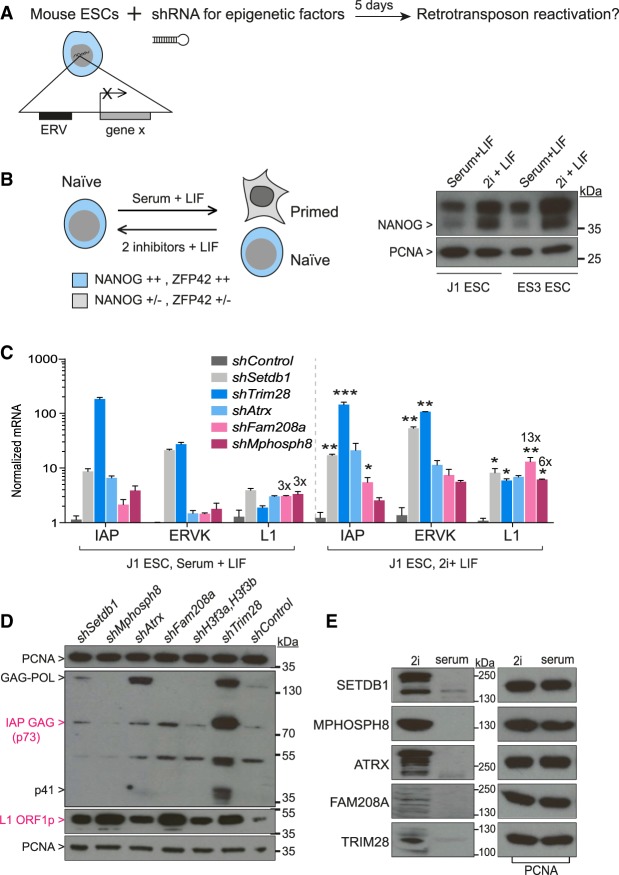
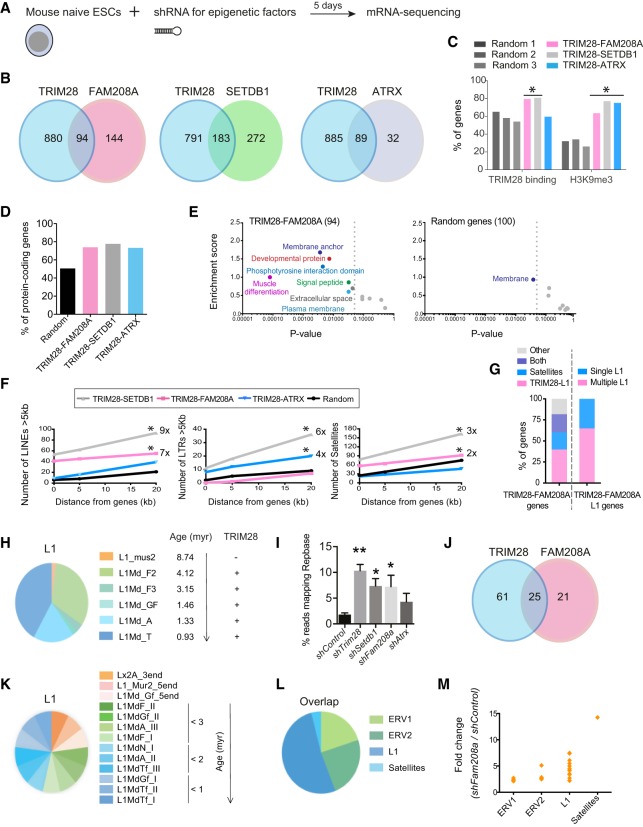
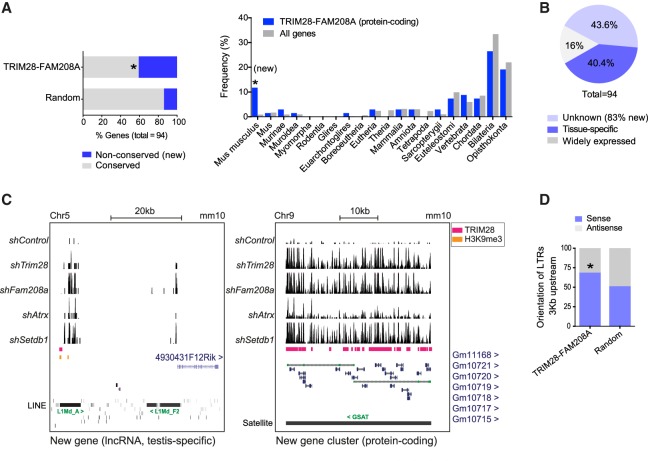
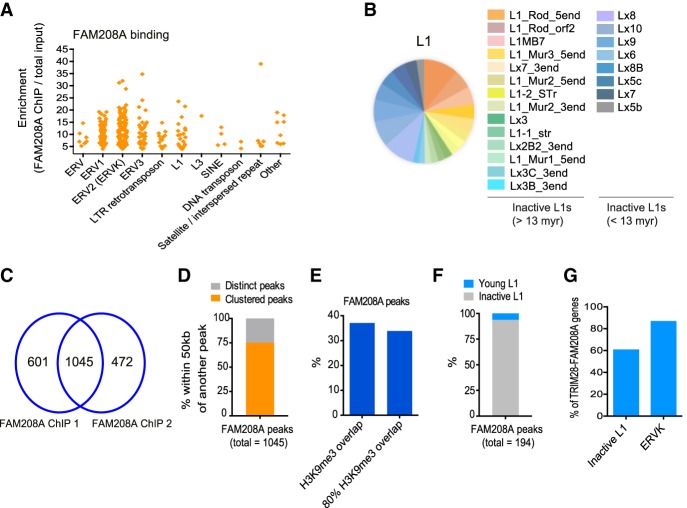
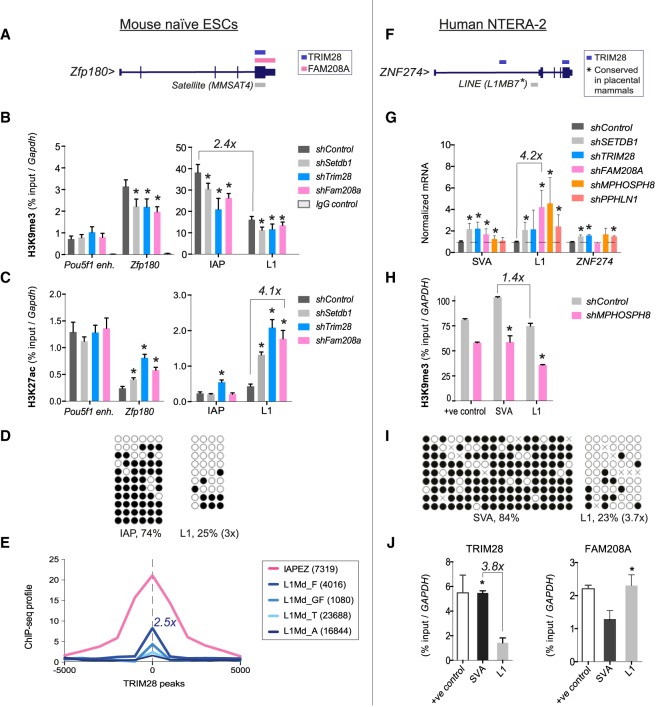
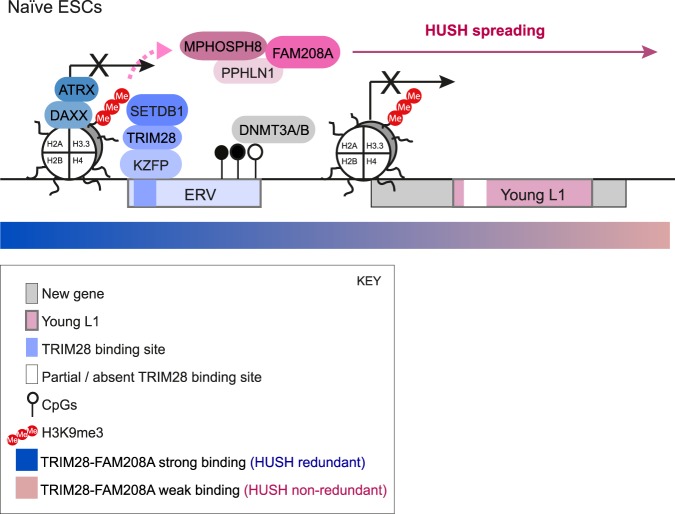
Retrotransposons encompass half of the human genome and contribute to the formation of heterochromatin, which provides nuclear structure and regulates gene expression. Here, we asked if the human silencing hub (HUSH) complex is necessary to silence retrotransposons and whether it collaborates with TRIM28 and the chromatin remodeler ATRX at specific genomic loci. We show that the HUSH complex contributes to de novo repression and DNA methylation of an SVA retrotransposon reporter. By using naïve versus primed mouse pluripotent stem cells, we reveal a critical role for the HUSH complex in naïve cells, implicating it in programming epigenetic marks in development. Although the HUSH component FAM208A binds to endogenous retroviruses (ERVs) and long interspersed element-1s (LINE-1s or L1s), it is mainly required to repress evolutionarily young L1s (mouse-specific lineages <5 million years old). TRIM28, in contrast, is necessary to repress both ERVs and young L1s. Genes co-repressed by TRIM28 and FAM208A are evolutionarily young, or exhibit tissue-specific expression, are enriched in young L1s, and display evidence for regulation through LTR promoters. Finally, we demonstrate that the HUSH complex is also required to repress L1 elements in human cells. Overall, these data indicate that the HUSH complex and TRIM28 co-repress young retrotransposons and new genes rewired by retrotransposon noncoding DNA.
© 2018 Robbez-Masson et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Genome surveillance by HUSH-mediated silencing of intronless mobile elements.Nature. 2022 Jan;601(7893):440-445. doi: 10.1038/s41586-021-04228-1. Epub 2021 Nov 18. Nature. 2022. PMID: 34794168 Free PMC article.
-
TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells.Genome Res. 2013 Mar;23(3):452-61. doi: 10.1101/gr.147678.112. Epub 2012 Dec 10. Genome Res. 2013. PMID: 23233547 Free PMC article.
-
TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells.Cell Rep. 2015 Jan 6;10(1):20-8. doi: 10.1016/j.celrep.2014.12.004. Epub 2014 Dec 24. Cell Rep. 2015. PMID: 25543143 Free PMC article.
-
Silencing of endogenous retroviruses by heterochromatin.Cell Mol Life Sci. 2017 Jun;74(11):2055-2065. doi: 10.1007/s00018-017-2454-8. Epub 2017 Feb 3. Cell Mol Life Sci. 2017. PMID: 28160052 Free PMC article. Review.
-
The sound of silence: mechanisms and implications of HUSH complex function.Trends Genet. 2023 Apr;39(4):251-267. doi: 10.1016/j.tig.2022.12.005. Epub 2023 Feb 6. Trends Genet. 2023. PMID: 36754727 Review.
Cited by
-
Human Endogenous Retrovirus Reactivation: Implications for Cancer Immunotherapy.Cancers (Basel). 2021 Apr 21;13(9):1999. doi: 10.3390/cancers13091999. Cancers (Basel). 2021. PMID: 33919186 Free PMC article. Review.
-
Tissue-specific usage of transposable element-derived promoters in mouse development.Genome Biol. 2020 Sep 28;21(1):255. doi: 10.1186/s13059-020-02164-3. Genome Biol. 2020. PMID: 32988383 Free PMC article.
-
Transcriptional Regulation of Endogenous Retroviruses and Their Misregulation in Human Diseases.Int J Mol Sci. 2022 Sep 4;23(17):10112. doi: 10.3390/ijms231710112. Int J Mol Sci. 2022. PMID: 36077510 Free PMC article. Review.
-
DNA methylation governs the sensitivity of repeats to restriction by the HUSH-MORC2 corepressor.Nat Commun. 2024 Aug 30;15(1):7534. doi: 10.1038/s41467-024-50765-4. Nat Commun. 2024. PMID: 39214989 Free PMC article.
-
Altered DNA methylation of TRIM13 in diabetic nephropathy suppresses mesangial collagen synthesis by promoting ubiquitination of CHOP.EBioMedicine. 2020 Jan;51:102582. doi: 10.1016/j.ebiom.2019.11.043. Epub 2020 Jan 2. EBioMedicine. 2020. PMID: 31901873 Free PMC article.
References
-
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc Ser B 57: 289–300.
-
- Brummelkamp TR, van Steensel B. 2015. GENE REGULATION. A HUSH for transgene expression. Science 348: 1433–1434. - PubMed
-
- Cammas F, Mark M, Dolle P, Dierich A, Chambon P, Losson R. 2000. Mice lacking the transcriptional corepressor TIF1β are defective in early postimplantation development. Development 127: 2955–2963. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous