

Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis
- PMID: 29686269
- PMCID: PMC5913300
- DOI: 10.1038/s41419-018-0469-1
Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis
Abstract
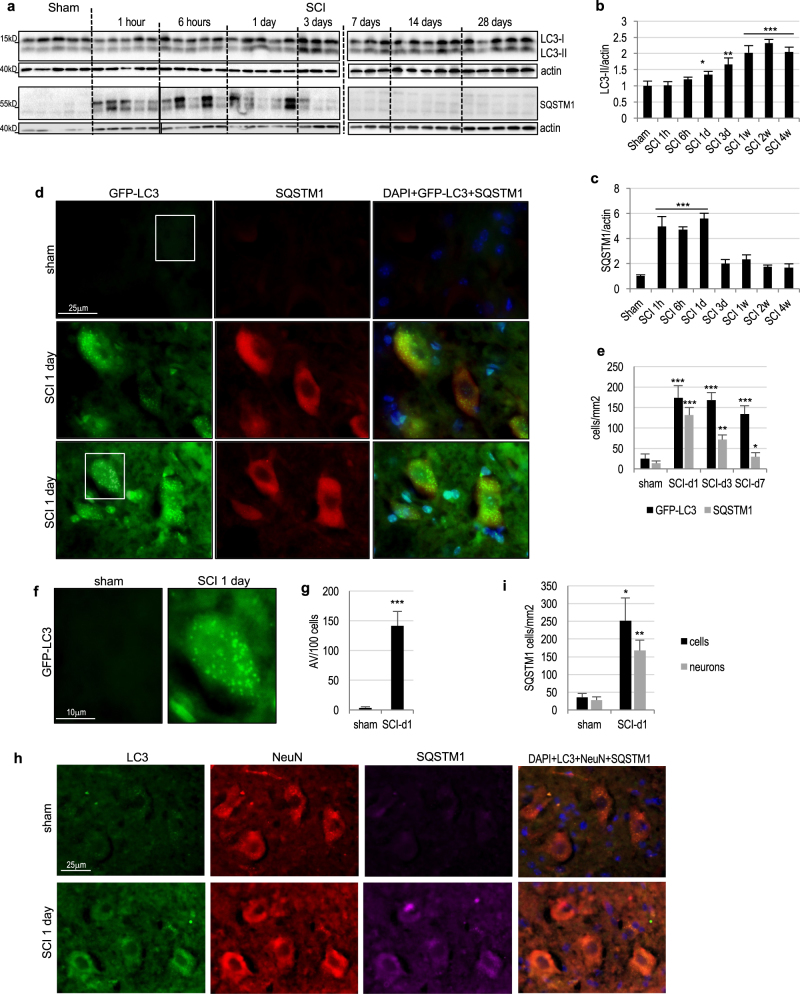
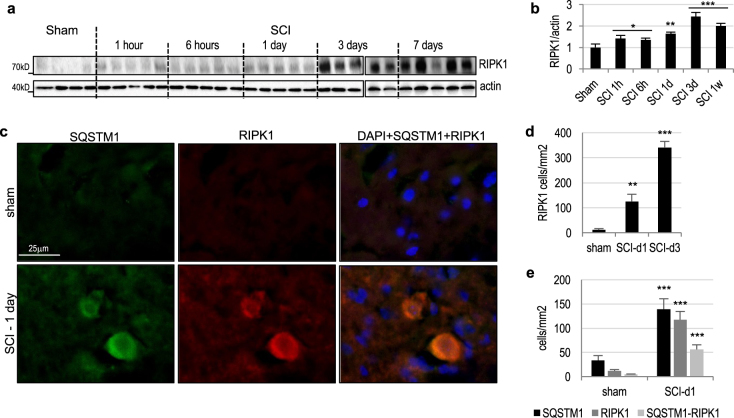
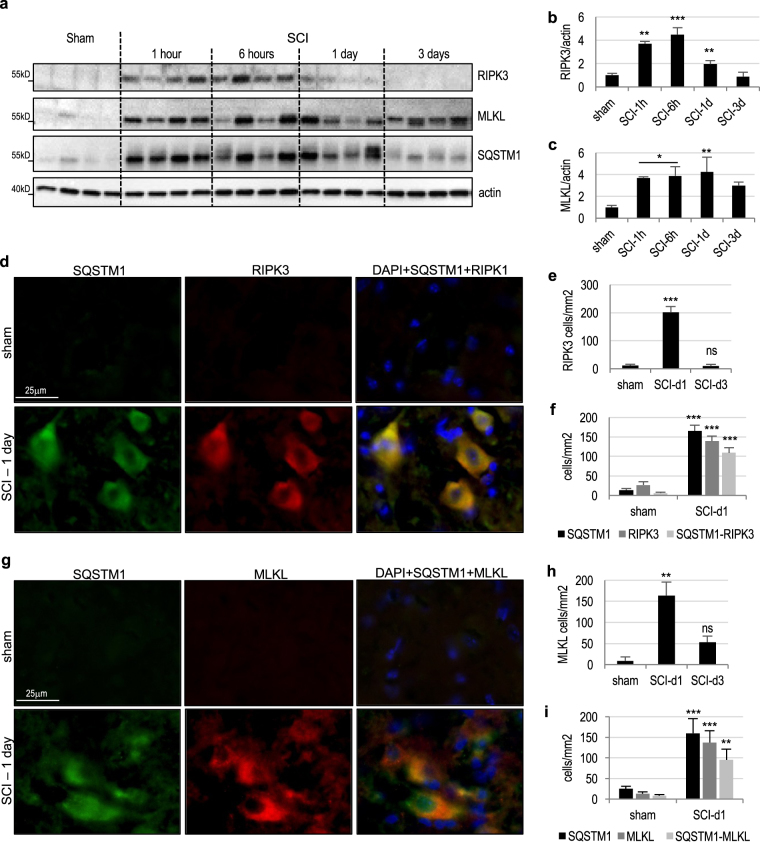
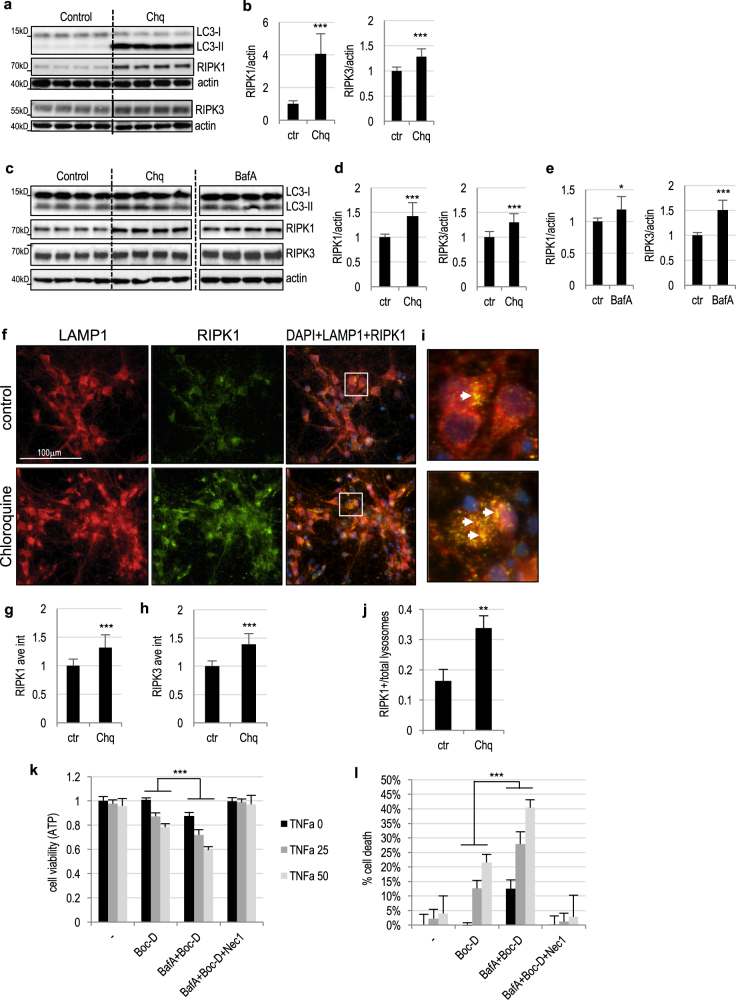
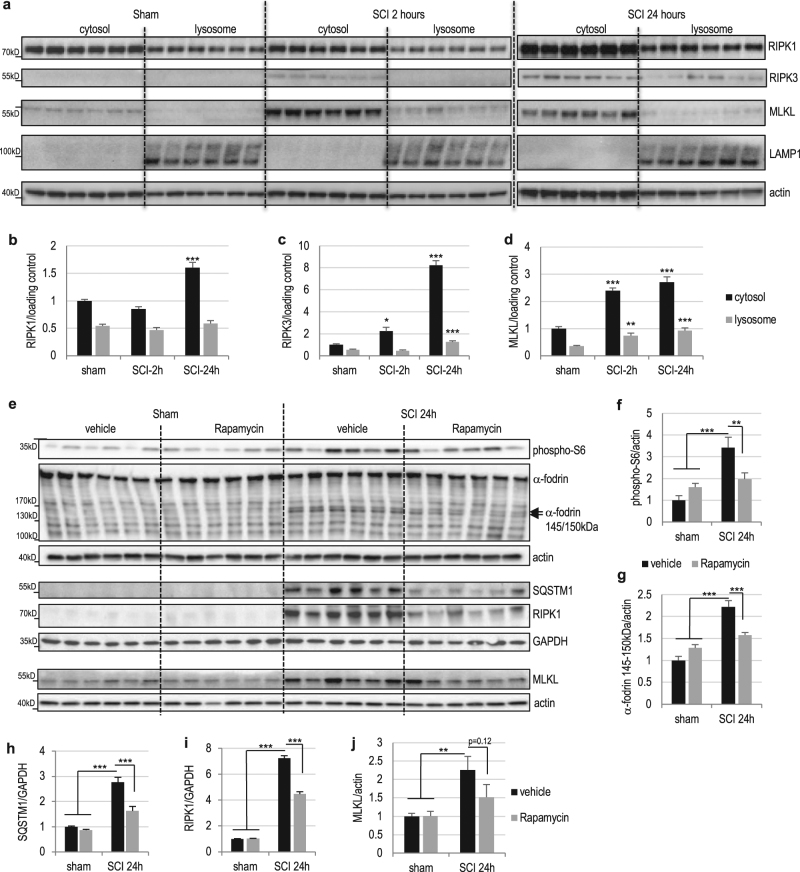
Necroptosis, a regulated necrosis pathway mediated by the receptor-interacting protein kinases 1 and 3 (RIPK1 and RIPK3), is induced following spinal cord injury (SCI) and thought to contribute to neuronal and glial cell death. However, mechanisms leading to activation of necroptosis after SCI remain unclear. We have previously shown that autophagy, a catabolic pathway facilitating degradation of cytoplasmic proteins and organelles in a lysosome-dependent manner, is inhibited following SCI in rats. Our current data confirm that inhibition of autophagy also occurs after thoracic contusive SCI in the mouse model, as indicated by accumulation of both the autophagosome marker, LC3-II and autophagy cargo protein, p62/SQSTM1. This was most pronounced in the ventral horn neurons and was caused by rapid inhibition of lysosomal function after SCI. Interestingly, RIPK1, RIPK3, and the necroptosis effector protein MLKL also rapidly accumulated after SCI and localized to neurons with disrupted autophagy, suggesting that these events may be related. To determine if lysosomal dysfunction could contribute to induction of necroptosis, we treated PC12 cells and primary rat cortical neurons with lysosomal inhibitors. This led to rapid accumulation of RIPK1 and RIPK3, confirming that they are normally degraded by the lysosomal pathway. In PC12 cells lysosomal inhibition also sensitized cells to necroptosis induced by tumor necrosis factor α (TNFα) and caspase inhibitor. Imaging studies confirmed that RIPK1 partially localized to lysosomes in both untreated and lysosomal inhibitor treated cells. Similarly, we detected presence of RIPK1, RIPK3 and MLKL in both cytosol and at lysosomes after SCI in vivo. Furthermore, stimulation of autophagy and lysosomal function with rapamycin treatment led to decreased accumulation of RIPK1 and attenuated cell death after SCI. These data suggest that lysosomal dysfunction after SCI may contribute to both inhibition of autophagy and sensitize cells to necroptosis by promoting RIPK1 and RIPK3 accumulation.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Similar articles
-
CAMK2/CaMKII activates MLKL in short-term starvation to facilitate autophagic flux.Autophagy. 2022 Apr;18(4):726-744. doi: 10.1080/15548627.2021.1954348. Epub 2021 Jul 20. Autophagy. 2022. PMID: 34282994 Free PMC article.
-
The neurotoxicant PCB-95 by increasing the neuronal transcriptional repressor REST down-regulates caspase-8 and increases Ripk1, Ripk3 and MLKL expression determining necroptotic neuronal death.Biochem Pharmacol. 2017 Oct 15;142:229-241. doi: 10.1016/j.bcp.2017.06.135. Epub 2017 Jul 1. Biochem Pharmacol. 2017. PMID: 28676433
-
cPLA2 activation contributes to lysosomal defects leading to impairment of autophagy after spinal cord injury.Cell Death Dis. 2019 Jul 11;10(7):531. doi: 10.1038/s41419-019-1764-1. Cell Death Dis. 2019. PMID: 31296844 Free PMC article.
-
RIPK1 and RIPK3: critical regulators of inflammation and cell death.Trends Cell Biol. 2015 Jun;25(6):347-53. doi: 10.1016/j.tcb.2015.01.001. Epub 2015 Feb 4. Trends Cell Biol. 2015. PMID: 25662614 Review.
-
The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis.Int Rev Cell Mol Biol. 2017;328:253-275. doi: 10.1016/bs.ircmb.2016.08.007. Epub 2016 Sep 22. Int Rev Cell Mol Biol. 2017. PMID: 28069136 Free PMC article. Review.
Cited by
-
GDF-11 Protects the Traumatically Injured Spinal Cord by Suppressing Pyroptosis and Necroptosis via TFE3-Mediated Autophagy Augmentation.Oxid Med Cell Longev. 2021 Oct 19;2021:8186877. doi: 10.1155/2021/8186877. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34712387 Free PMC article.
-
The role of lysosome in regulated necrosis.Acta Pharm Sin B. 2020 Oct;10(10):1880-1903. doi: 10.1016/j.apsb.2020.07.003. Epub 2020 Jul 13. Acta Pharm Sin B. 2020. PMID: 33163342 Free PMC article. Review.
-
Role of necroptosis in traumatic brain and spinal cord injuries.J Adv Res. 2022 Sep;40:125-134. doi: 10.1016/j.jare.2021.12.002. Epub 2021 Dec 22. J Adv Res. 2022. PMID: 36100321 Free PMC article. Review.
-
Research Progress on Micro(nano)plastic-Induced Programmed Cell Death Associated with Disease Risks.Toxics. 2024 Jul 5;12(7):493. doi: 10.3390/toxics12070493. Toxics. 2024. PMID: 39058145 Free PMC article. Review.
-
3,4-Dimethoxychalcone, a caloric restriction mimetic, enhances TFEB-mediated autophagy and alleviates pyroptosis and necroptosis after spinal cord injury.Theranostics. 2023 Jan 1;13(2):810-832. doi: 10.7150/thno.78370. eCollection 2023. Theranostics. 2023. PMID: 36632211 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
