

Two distinct DNA sequences recognized by transcription factors represent enthalpy and entropy optima
- PMID: 29638214
- PMCID: PMC5896879
- DOI: 10.7554/eLife.32963
Two distinct DNA sequences recognized by transcription factors represent enthalpy and entropy optima
Abstract
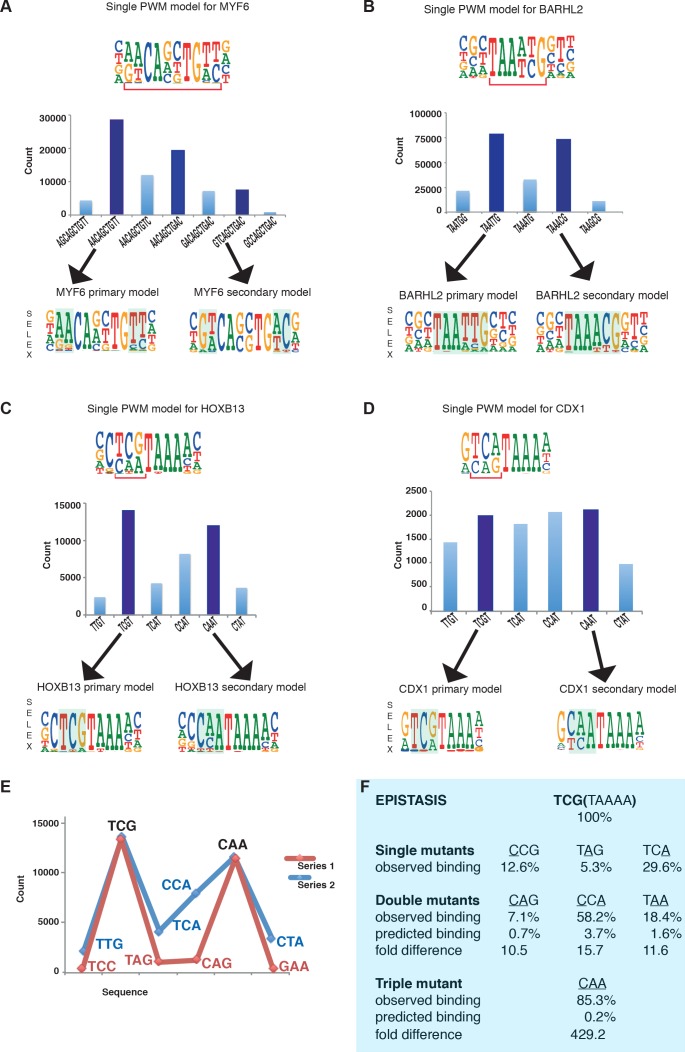
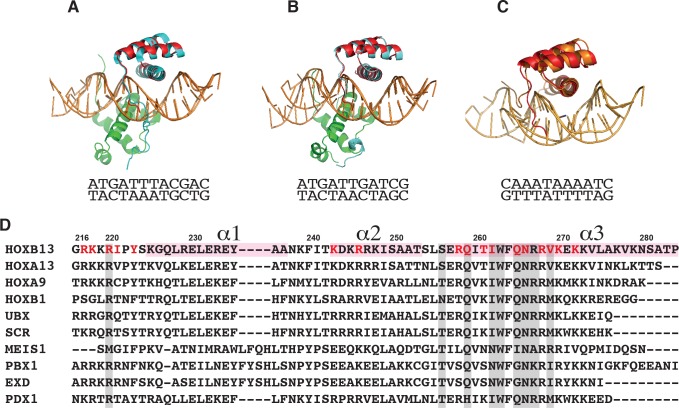
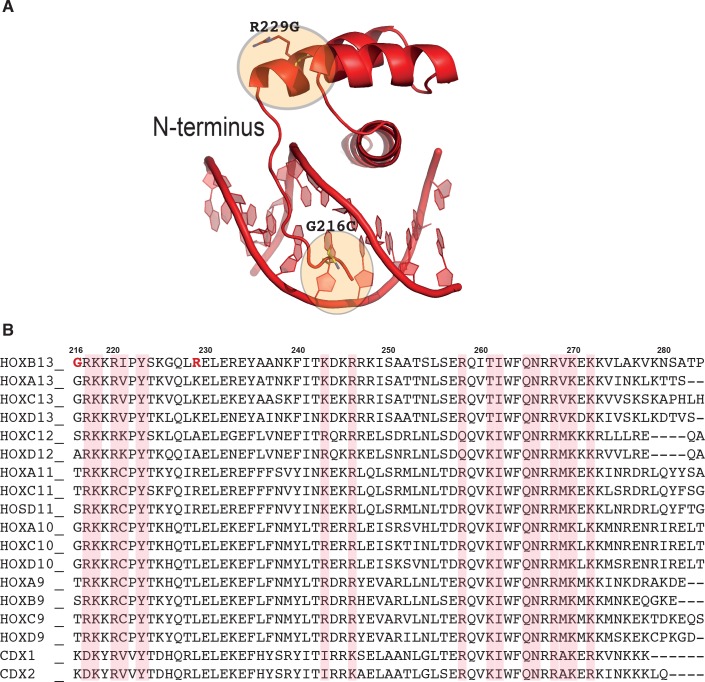
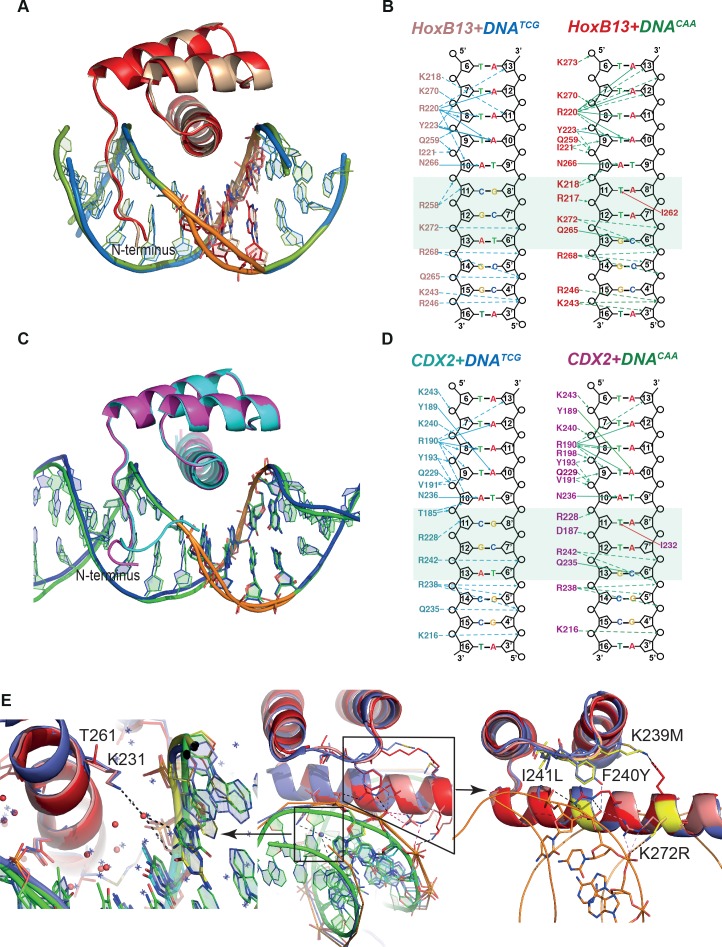
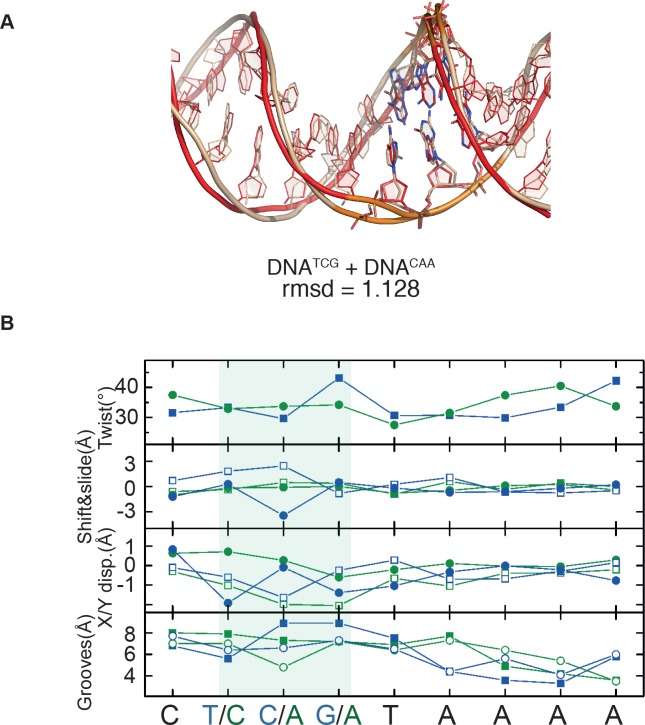
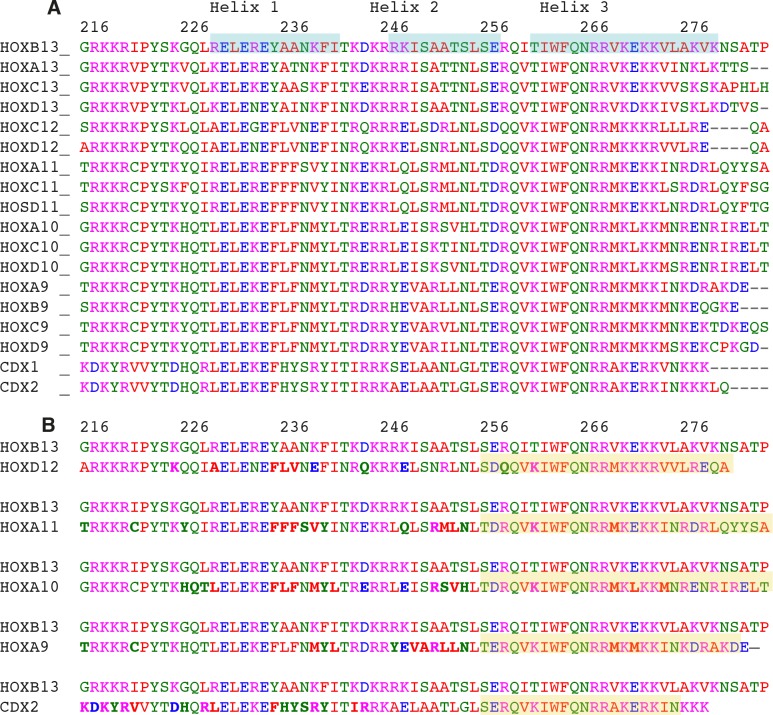
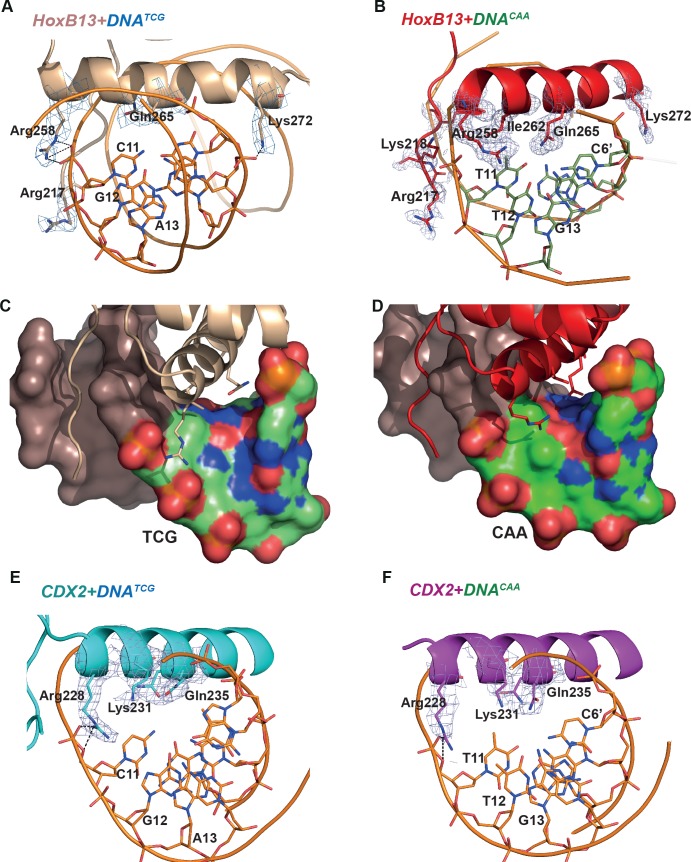
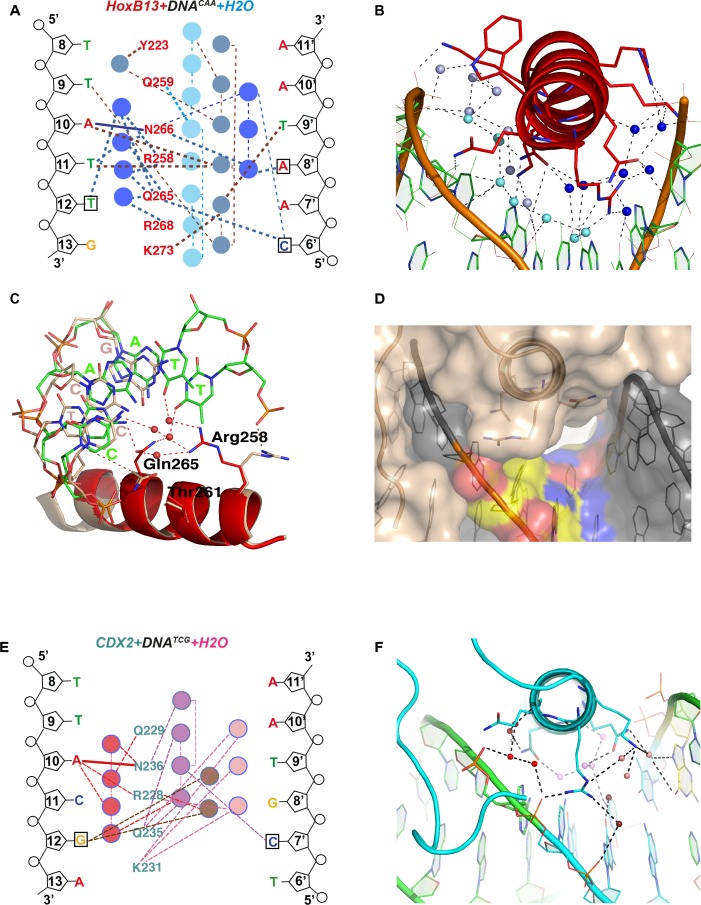
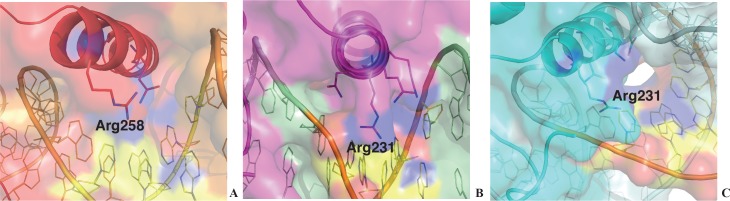
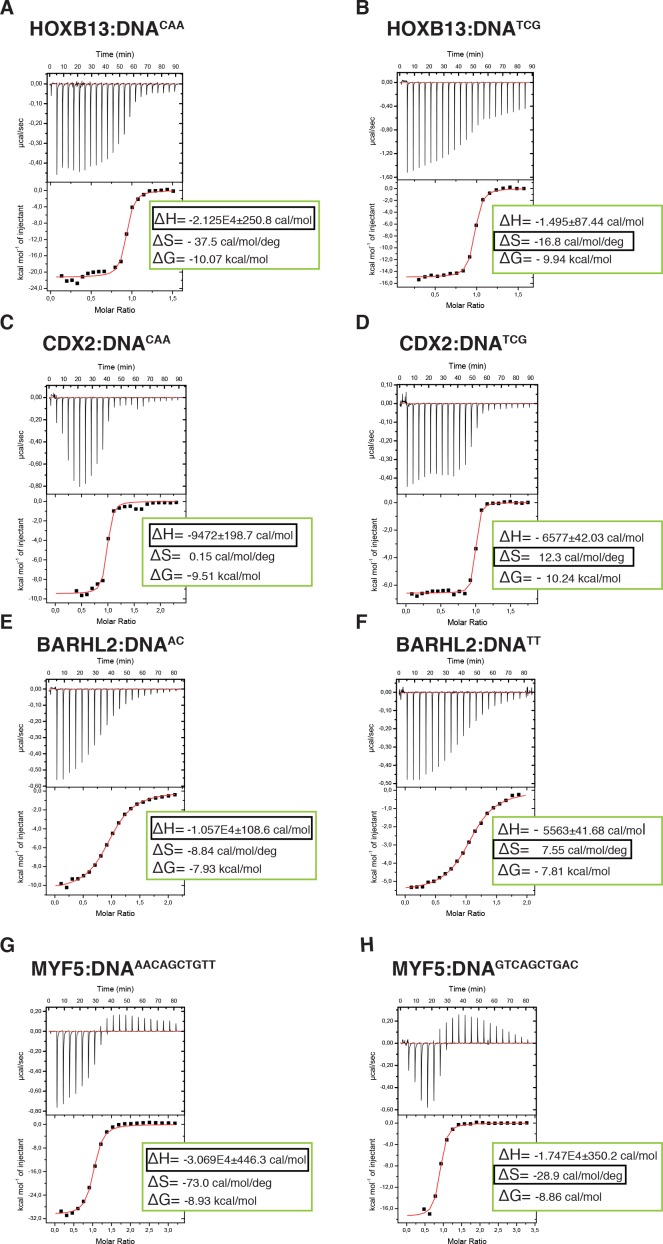
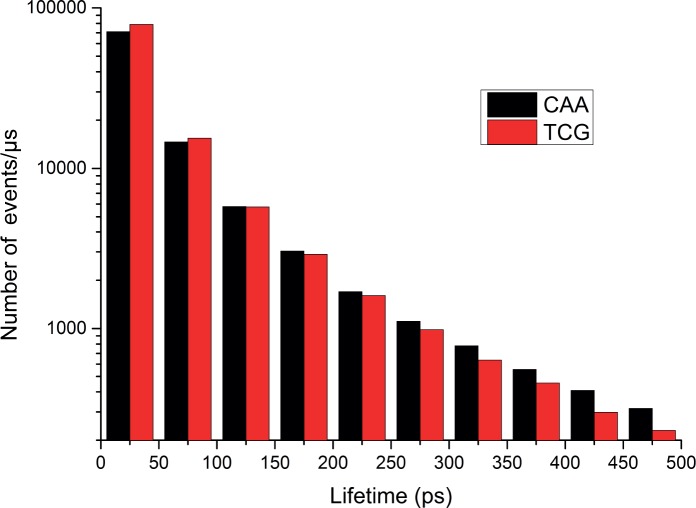
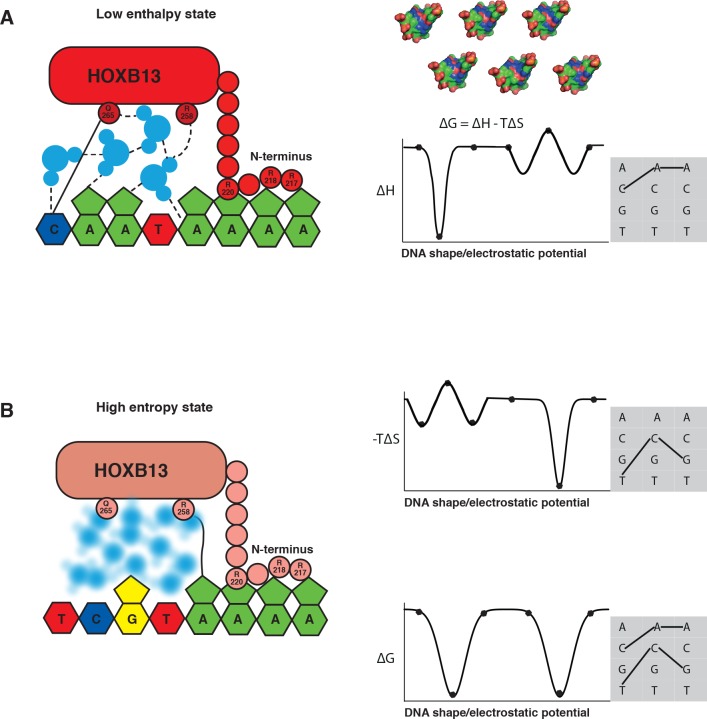
Most transcription factors (TFs) can bind to a population of sequences closely related to a single optimal site. However, some TFs can bind to two distinct sequences that represent two local optima in the Gibbs free energy of binding (ΔG). To determine the molecular mechanism behind this effect, we solved the structures of human HOXB13 and CDX2 bound to their two optimal DNA sequences, CAATAAA and TCGTAAA. Thermodynamic analyses by isothermal titration calorimetry revealed that both sites were bound with similar ΔG. However, the interaction with the CAA sequence was driven by change in enthalpy (ΔH), whereas the TCG site was bound with similar affinity due to smaller loss of entropy (ΔS). This thermodynamic mechanism that leads to at least two local optima likely affects many macromolecular interactions, as ΔG depends on two partially independent variables ΔH and ΔS according to the central equation of thermodynamics, ΔG = ΔH - TΔS.
Keywords: DNA recognition; biochemistry; chemical biology; enthalpy; entropy; epistasis; human; molecular biophysics; structural biology; thermodynamics; transcription factors.
© 2018, Morgunova et al.
Conflict of interest statement
EM, YY, PD, AJ, FZ, AP, YX, LN, JT No competing interests declared
Figures
Similar articles
-
Structural and thermodynamic strategies for site-specific DNA binding proteins.Structure. 2000 Oct 15;8(10):1015-23. doi: 10.1016/s0969-2126(00)00501-3. Structure. 2000. PMID: 11080623
-
Comparative thermodynamics for monomer and dimer sequence-dependent binding of a heterocyclic dication in the DNA minor groove.J Mol Biol. 2002 Mar 29;317(3):361-74. doi: 10.1006/jmbi.2002.5433. J Mol Biol. 2002. PMID: 11922670
-
Intrinsic Thermodynamics of Protein-Ligand Binding by Isothermal Titration Calorimetry as Aid to Drug Design.Methods Mol Biol. 2019;1964:61-74. doi: 10.1007/978-1-4939-9179-2_5. Methods Mol Biol. 2019. PMID: 30929235
-
Thermodynamic study of aptamers binding to their target proteins.Biochimie. 2018 Feb;145:91-97. doi: 10.1016/j.biochi.2017.10.010. Epub 2017 Oct 18. Biochimie. 2018. PMID: 29054802 Review.
-
The Enthalpy-entropy Compensation Phenomenon. Limitations for the Use of Some Basic Thermodynamic Equations.Curr Protein Pept Sci. 2018;19(11):1088-1091. doi: 10.2174/1389203719666180521092615. Curr Protein Pept Sci. 2018. PMID: 29779476 Free PMC article. Review.
Cited by
-
Enthalpy and entropy synergistic regulation-based programmable DNA motifs for biosensing and information encryption.Sci Adv. 2023 May 19;9(20):eadf5868. doi: 10.1126/sciadv.adf5868. Epub 2023 May 17. Sci Adv. 2023. PMID: 37196083 Free PMC article.
-
The bZIP mutant CEBPB (V285A) has sequence specific DNA binding propensities similar to CREB1.Biochim Biophys Acta Gene Regul Mech. 2019 Apr;1862(4):486-492. doi: 10.1016/j.bbagrm.2019.02.002. Epub 2019 Feb 27. Biochim Biophys Acta Gene Regul Mech. 2019. PMID: 30825655 Free PMC article.
-
DNA binding analysis of rare variants in homeodomains reveals homeodomain specificity-determining residues.Nat Commun. 2024 Apr 10;15(1):3110. doi: 10.1038/s41467-024-47396-0. Nat Commun. 2024. PMID: 38600112 Free PMC article.
-
Energy landscape reshaped by strain-specific mutations underlies epistasis in NS1 evolution of influenza A virus.Nat Commun. 2022 Oct 1;13(1):5775. doi: 10.1038/s41467-022-33554-9. Nat Commun. 2022. PMID: 36182933 Free PMC article.
-
Noncanonical binding of transcription factors: time to revisit specificity?Mol Biol Cell. 2023 Aug 1;34(9):pe4. doi: 10.1091/mbc.E22-08-0325. Mol Biol Cell. 2023. PMID: 37486893 Free PMC article. Review.
References
-
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. - DOI - PMC - PubMed
-
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallographica Section D Biological Crystallography. 2012;68:352–367. doi: 10.1107/S0907444912001308. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
