

A CLK3-HMGA2 Alternative Splicing Axis Impacts Human Hematopoietic Stem Cell Molecular Identity throughout Development
- PMID: 29625070
- PMCID: PMC5957284
- DOI: 10.1016/j.stem.2018.03.012
A CLK3-HMGA2 Alternative Splicing Axis Impacts Human Hematopoietic Stem Cell Molecular Identity throughout Development
Abstract
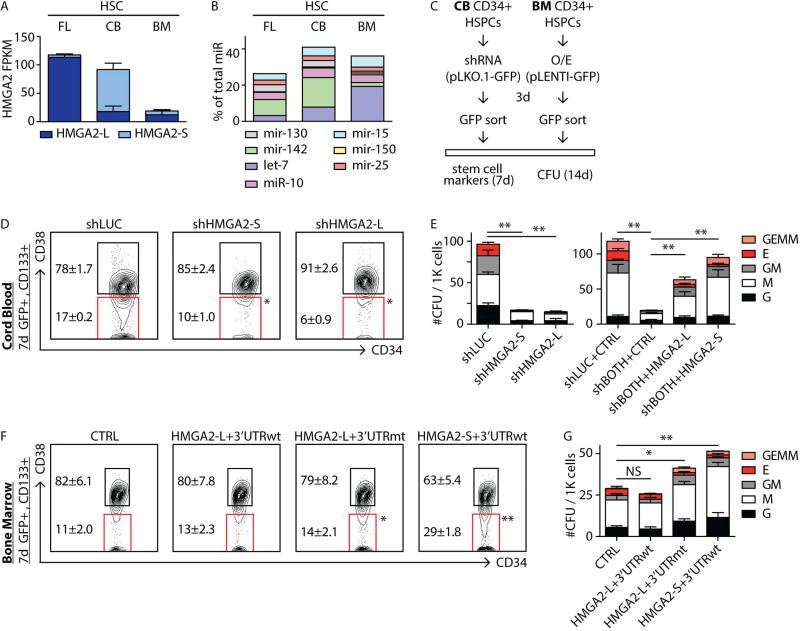
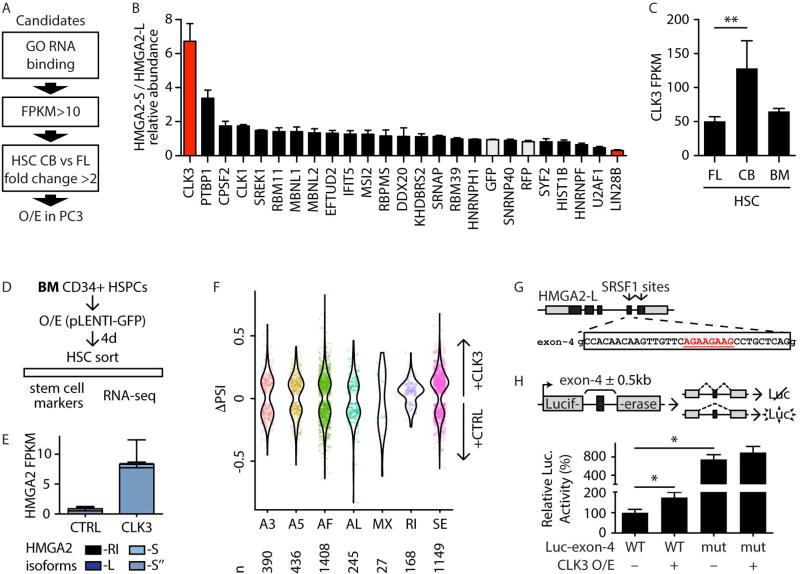
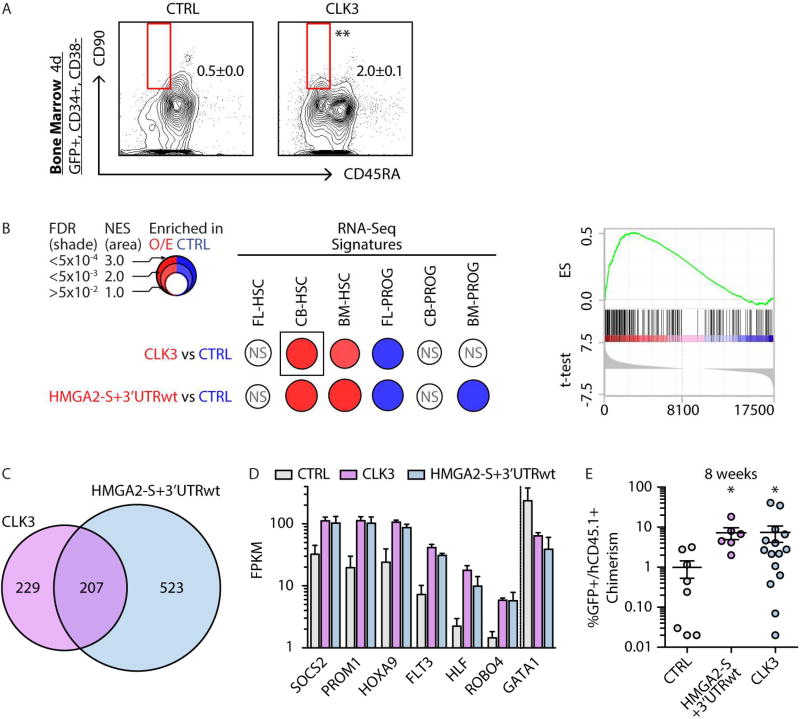
While gene expression dynamics have been extensively cataloged during hematopoietic differentiation in the adult, less is known about transcriptome diversity of human hematopoietic stem cells (HSCs) during development. To characterize transcriptional and post-transcriptional changes in HSCs during development, we leveraged high-throughput genomic approaches to profile miRNAs, lincRNAs, and mRNAs. Our findings indicate that HSCs manifest distinct alternative splicing patterns in key hematopoietic regulators. Detailed analysis of the splicing dynamics and function of one such regulator, HMGA2, identified an alternative isoform that escapes miRNA-mediated targeting. We further identified the splicing kinase CLK3 that, by regulating HMGA2 splicing, preserves HMGA2 function in the setting of an increase in let-7 miRNA levels, delineating how CLK3 and HMGA2 form a functional axis that influences HSC properties during development. Collectively, our study highlights molecular mechanisms by which alternative splicing and miRNA-mediated post-transcriptional regulation impact the molecular identity and stage-specific developmental features of human HSCs.
Keywords: CLK3; HMGA2; RNA-seq; SRSF1; alternative splicing; human hematopoietic stem cells.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
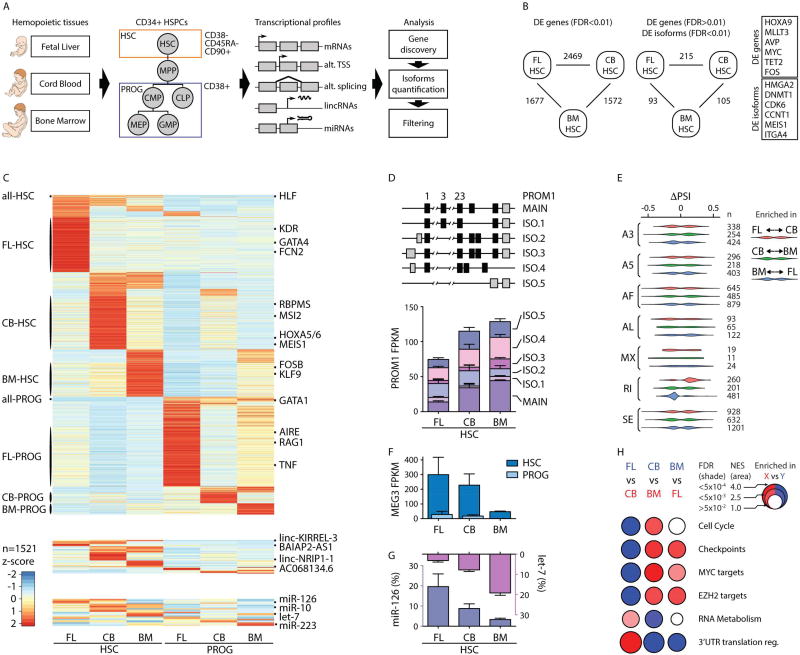
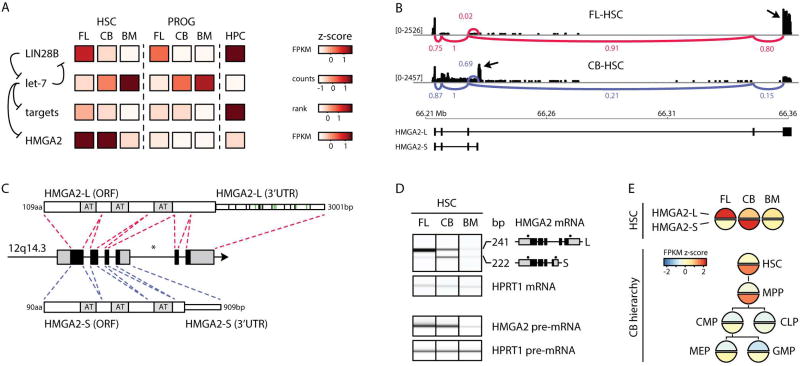
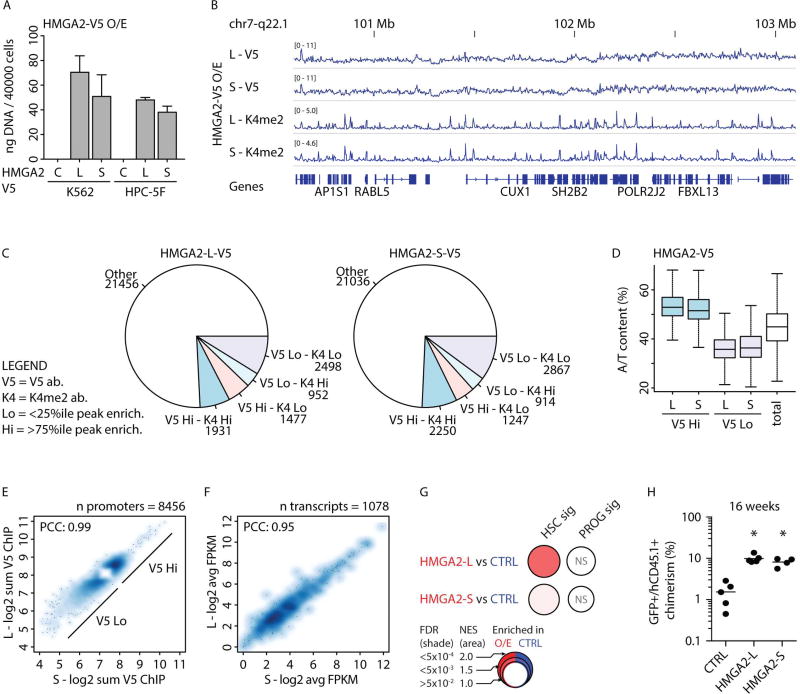
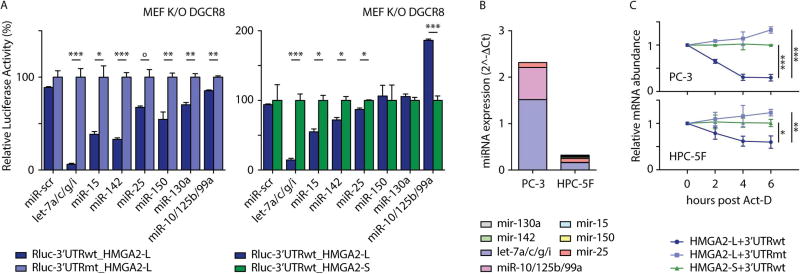
Comment in
-
Role of alternative splicing in hematopoietic stem cells during development.Stem Cell Investig. 2018 Aug 23;5:26. doi: 10.21037/sci.2018.08.02. eCollection 2018. Stem Cell Investig. 2018. PMID: 30221171 Free PMC article. No abstract available.
Similar articles
-
The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells.Nat Cell Biol. 2013 Aug;15(8):916-25. doi: 10.1038/ncb2783. Epub 2013 Jun 30. Nat Cell Biol. 2013. PMID: 23811688
-
Hypoxia leads to significant changes in alternative splicing and elevated expression of CLK splice factor kinases in PC3 prostate cancer cells.BMC Cancer. 2018 Apr 2;18(1):355. doi: 10.1186/s12885-018-4227-7. BMC Cancer. 2018. PMID: 29606096 Free PMC article.
-
Chromatin modifier Hmga2 promotes adult hematopoietic stem cell function and blood regeneration in stress conditions.EMBO J. 2024 Jul;43(13):2661-2684. doi: 10.1038/s44318-024-00122-4. Epub 2024 May 29. EMBO J. 2024. PMID: 38811851 Free PMC article.
-
Developmental changes in hematopoietic stem cell properties.Exp Mol Med. 2013 Nov 15;45(11):e55. doi: 10.1038/emm.2013.98. Exp Mol Med. 2013. PMID: 24232254 Free PMC article. Review.
-
Post-transcriptional regulation in hematopoiesis: RNA binding proteins take control 1.Biochem Cell Biol. 2019 Feb;97(1):10-20. doi: 10.1139/bcb-2017-0310. Epub 2018 Jun 13. Biochem Cell Biol. 2019. PMID: 29898370 Review.
Cited by
-
Genome-scale analysis of Arabidopsis splicing-related protein kinase families reveals roles in abiotic stress adaptation.BMC Plant Biol. 2022 Oct 22;22(1):496. doi: 10.1186/s12870-022-03870-9. BMC Plant Biol. 2022. PMID: 36273172 Free PMC article.
-
Manganese dioxide nanosheets: from preparation to biomedical applications.Int J Nanomedicine. 2019 Jul 3;14:4781-4800. doi: 10.2147/IJN.S207666. eCollection 2019. Int J Nanomedicine. 2019. PMID: 31308658 Free PMC article. Review.
-
Cdc2-like kinases: structure, biological function, and therapeutic targets for diseases.Signal Transduct Target Ther. 2023 Apr 7;8(1):148. doi: 10.1038/s41392-023-01409-4. Signal Transduct Target Ther. 2023. PMID: 37029108 Free PMC article. Review.
-
RNA binding protein-directed control of leukemic stem cell evolution and function.Hemasphere. 2024 Aug 22;8(8):e116. doi: 10.1002/hem3.116. eCollection 2024 Aug. Hemasphere. 2024. PMID: 39175825 Free PMC article. Review.
-
Role of alternative splicing in hematopoietic stem cells during development.Stem Cell Investig. 2018 Aug 23;5:26. doi: 10.21037/sci.2018.08.02. eCollection 2018. Stem Cell Investig. 2018. PMID: 30221171 Free PMC article. No abstract available.
References
-
- Babovic S, Eaves CJ. Hierarchical organization of fetal and adult hematopoietic stem cells. Exp. Cell Res. 2014;329:185–191. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
